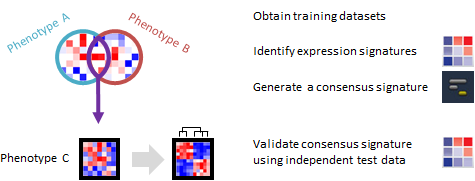
Do phenotypically different expression datasets share a common signature? Can the signature distinguish phenotypes in an independent dataset?
This recipe provides one method for identifying a consensus gene signature from a training set of several phenotypically distinct gene expression dataset. The recipe then validates the ability of the consensus signature to accurately distinguish phenotypes by using an independent test gene expression dataset. An example use case of this recipe is when an investigator may want to develop a gene expression signature to predict a specific phenotype, such as cancer or another disease.
Background information: What is a consensus gene expression signature?
A gene expression signature is the pattern of expression in a specific group of genes, usually ones that are related by function, position or other biological process. A consensus gene signature is an expression pattern for a specific group of genes, which is shared among different samples or across different phenotypes. For example, a group of genes regulating immune response could be similarly up-regulated during many different, unrelated infections. There are several types of consensus signatures; those that can be derived from gene expression data are called transcriptional consensus signatures. Consensus signatures can be created by overlapping individual gene signatures derived from multiple datasets. Compared to individual gene expression signatures, consensus signatures may be more accurate at distinguishing different phenotypes, such as diseased vs. normal samples.
Use case: Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition (Puissant et al. , Cancer Discov. 2013).
This study analyzed gene expression data generated from primary neuroblastoma tumors of two genetic classes: tumors harboring MYCN amplification (“MYCN amplified”) and tumors without MYCN amplification (“MYCN non-amplified”). MYCN amplified neuroblastoma is exquisitely dependent on the bromodomain and extra-terminal (BET) family of proteins. As such, treatment of MYCN amplified cell lines or tumors with JQ1, a small-molecule inhibitor of BET proteins, leads to dramatic transcriptional changes and induces cell death.
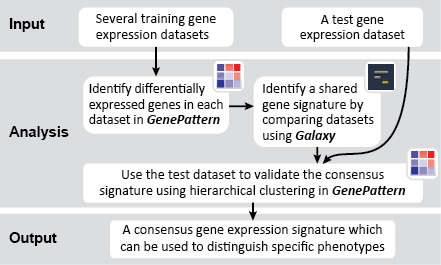
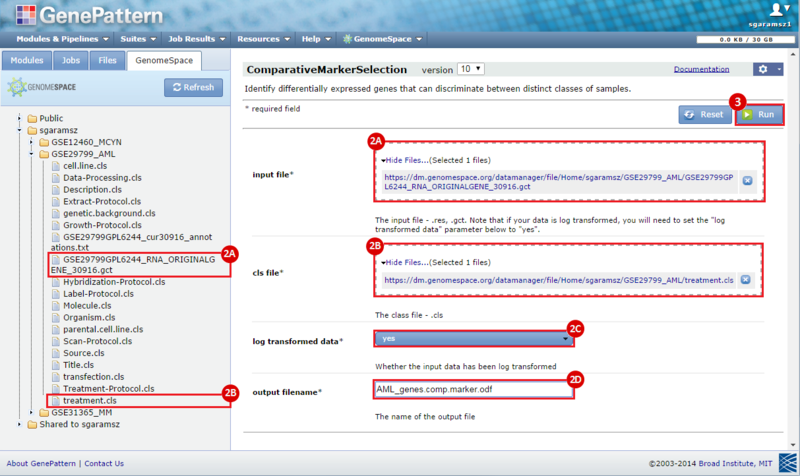
To identify a consensus signature to predict sensitivity to JQ1 treatment, two training datasets and one test dataset were used. The training dataset included acute myeloid leukemia (AML) and a multiple myeloid leukemia (MM) cell lines, which had been treated with either DMSO (control) or with JQ1 (treatment). The test dataset included MYCN amplified and MYCN nonamplified neuroblastoma primary tumor samples. GenePattern was used to analyze the AML and MM cell lines; for each dataset, a gene expression signature was derived to identify JQ1 response in the cell line. Using Galaxy, the two signatures were then overlapped to determine the consensus signature between the two phenotypes.
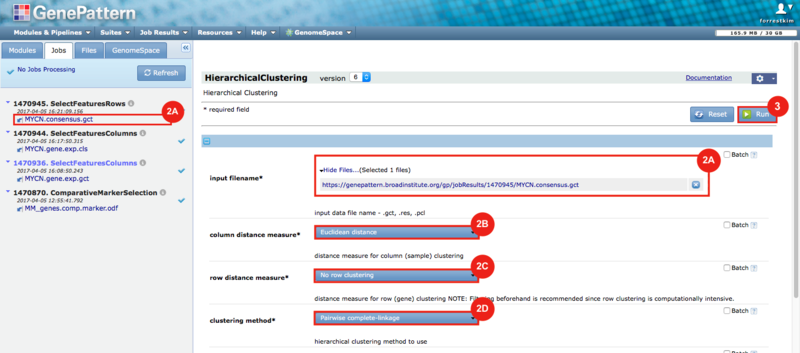
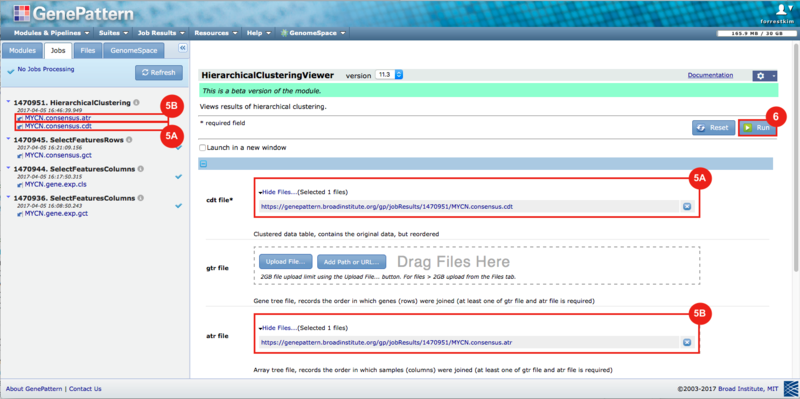
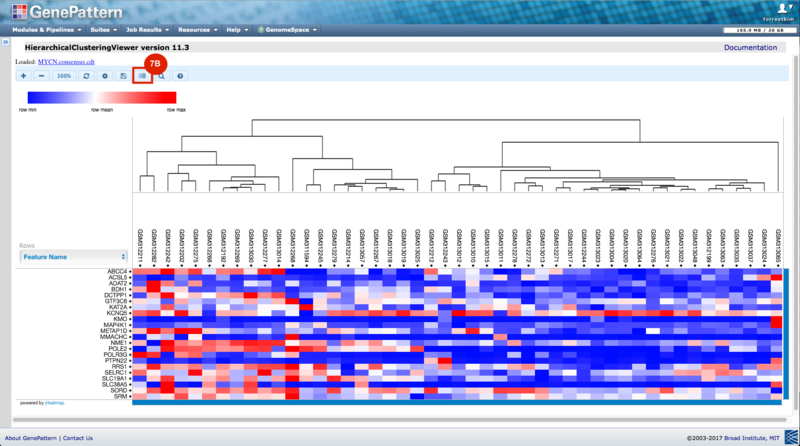
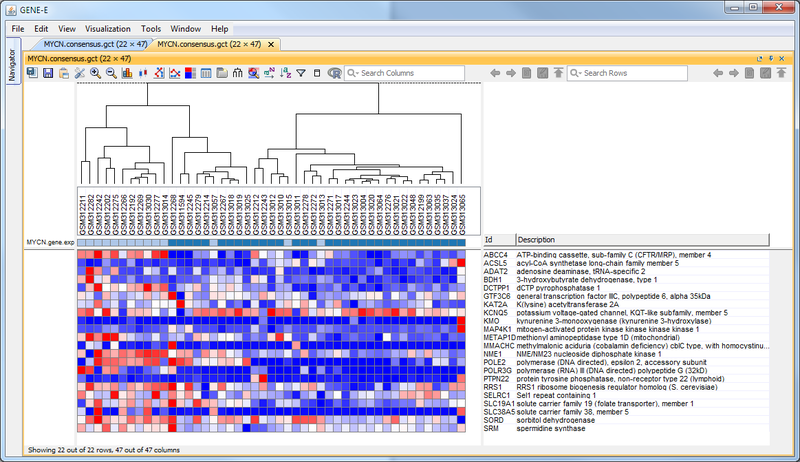
GenePattern was used to validate the ability of this JQ1-associated consensus signature to differentiate between phenotypes, by using the signature to hierarchically cluster the test dataset (neuroblastoma). Since the MYCN amplified and MYCN non-amplified neuroblastoma samples should have differing expression profiles, it was hypothesized that the consensus signature would be able to separate the samples by phenotype. Indeed, the consensus signature was able to cluster the MYCN-amplified and MYCN-nonamplified samples separately, revealing that the consensus signature accurately distinguishes the sensitivity-to-JQ1 phenotype.
To complete this recipe, we will need several gene expression datasets:
Public > RecipeData > ExpressionData > GSE29799_AML: a JQ1-treated acute myeloid leukemia (AML) expression dataset (6 samples)Public > RecipeData > ExpressionData > GSE31365_MM: a JQ1-treated multiple myeloma (MM) expression dataset (12 samples)Public > RecipeData > ExpressionData > GSE12460_NB: a neuroblastoma (NB) expression dataset which has MCYN-amplified and MYCN-nonamplified samples (64 samples)

 GenomeSpace.org
GenomeSpace.org


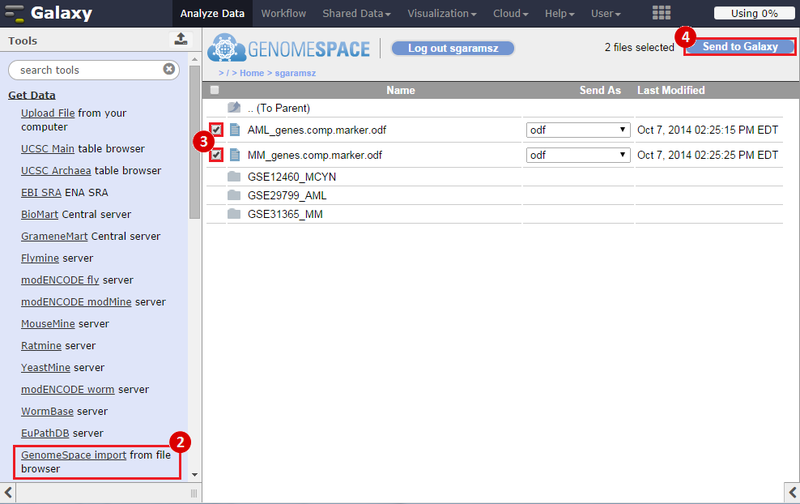
 icon in the upper right corner to import the workflow.
icon in the upper right corner to import the workflow.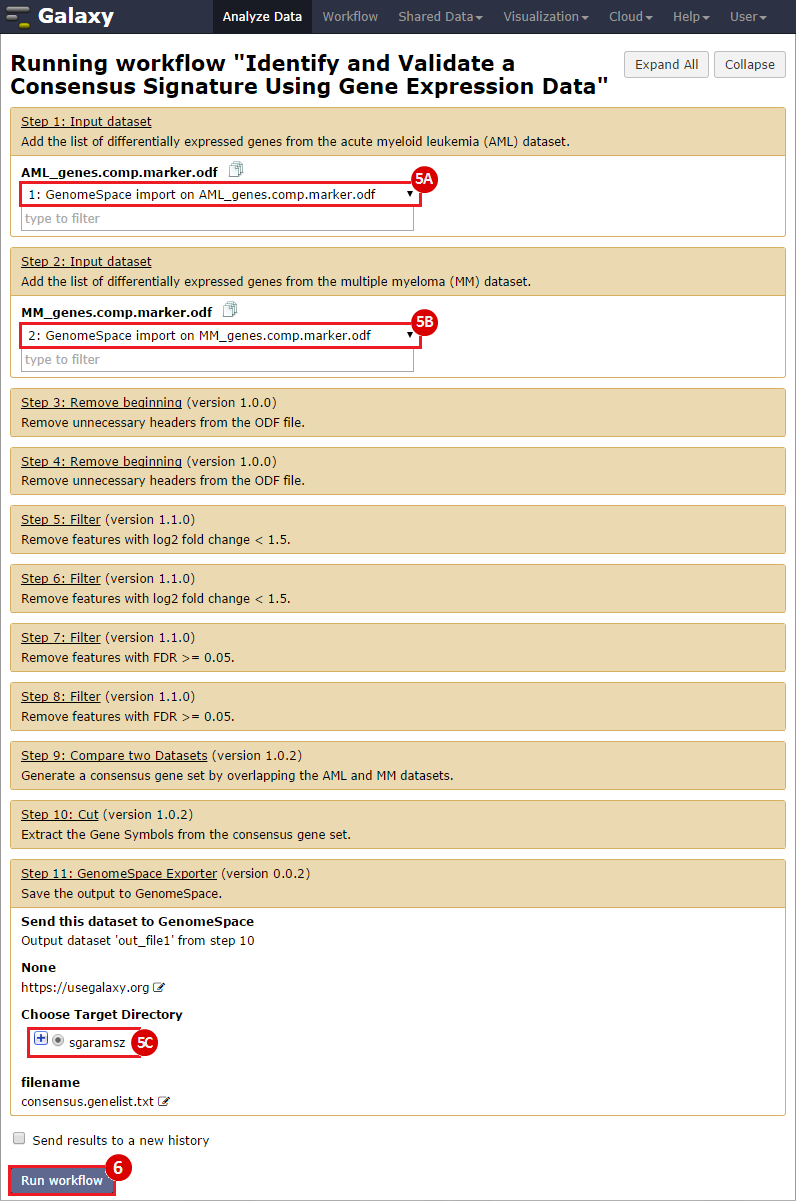
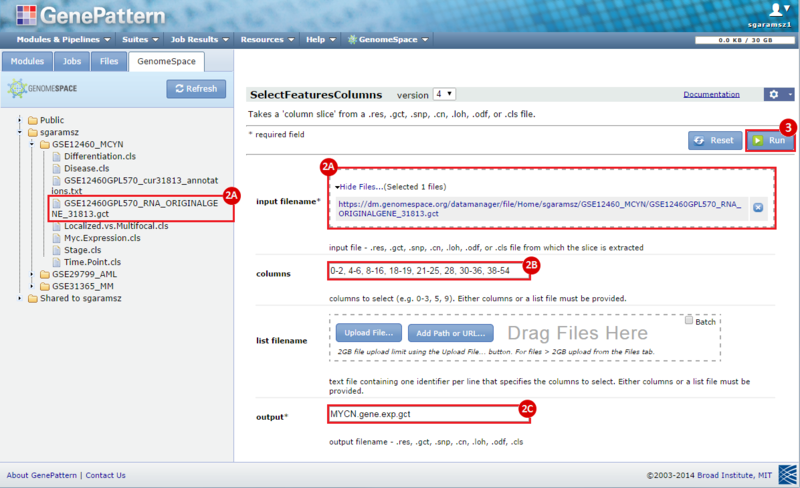
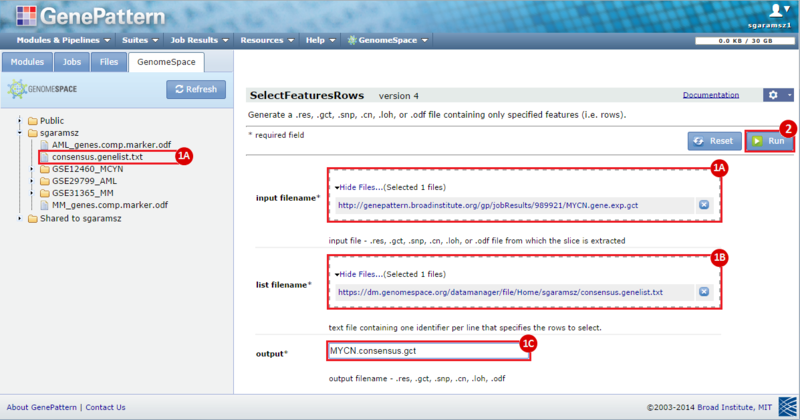
 next to the input filename parameter to remove the GCT file from the module. Then, use the GenomeSpace tab to navigate to the GSE12460_MYCN directory:
next to the input filename parameter to remove the GCT file from the module. Then, use the GenomeSpace tab to navigate to the GSE12460_MYCN directory: