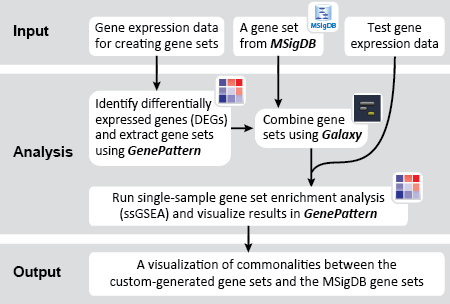
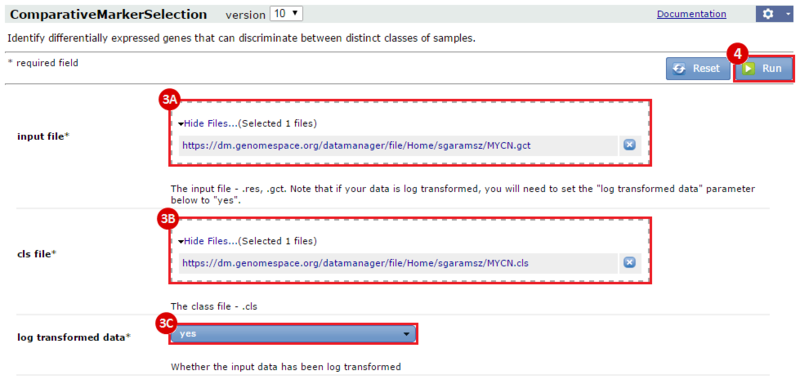
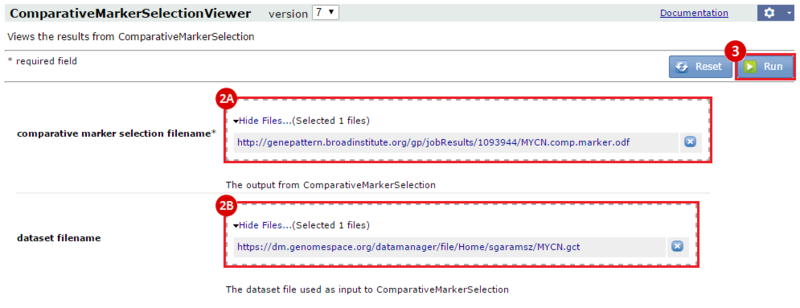
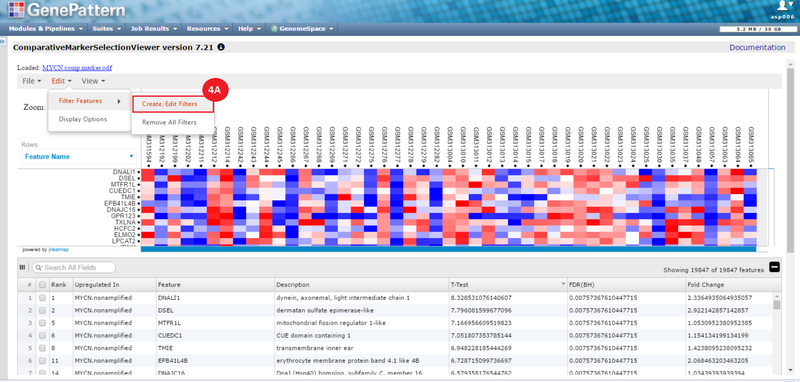
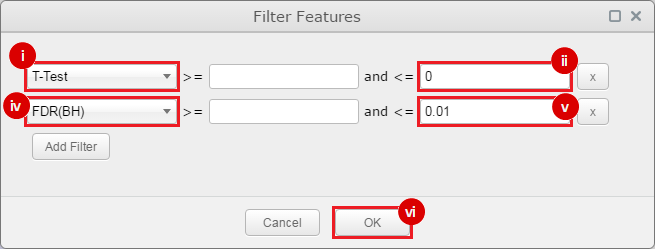
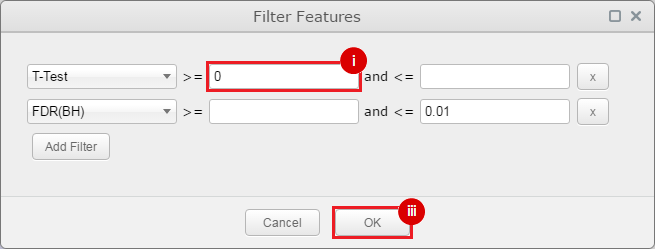
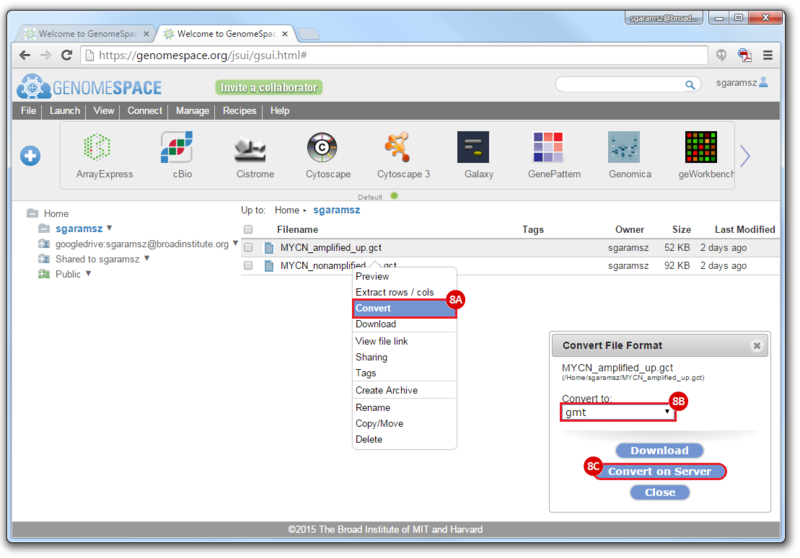
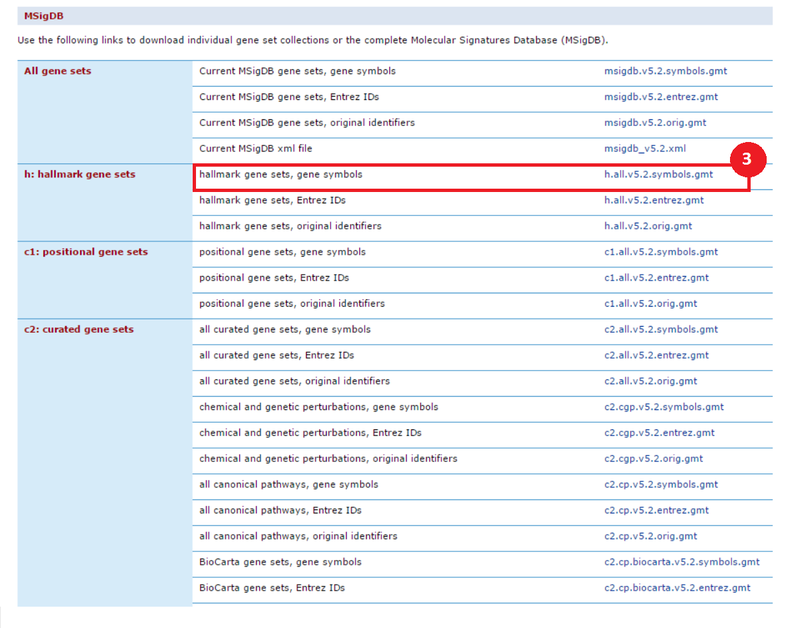
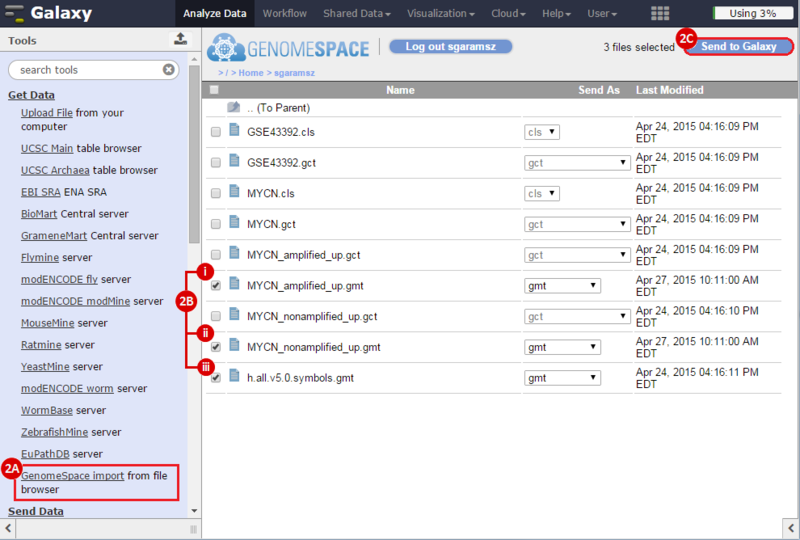
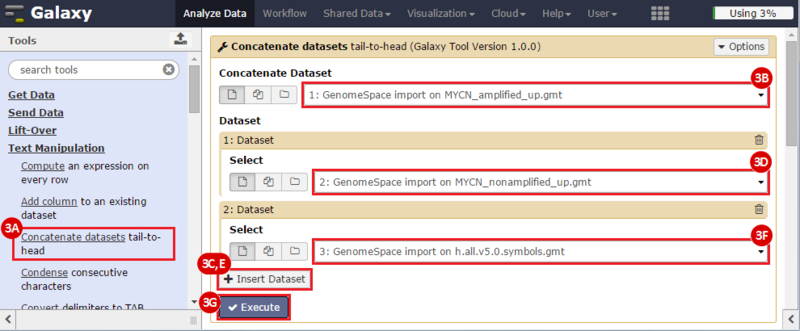
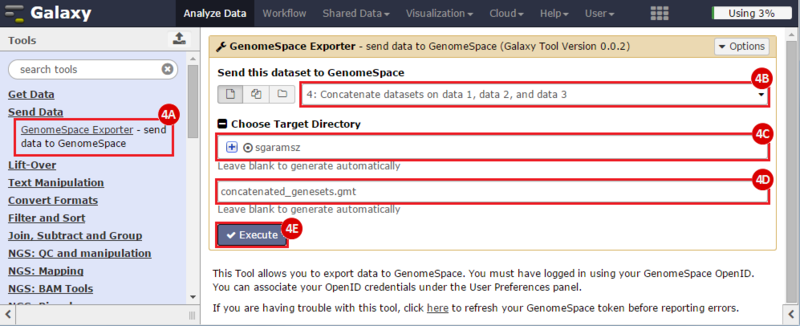
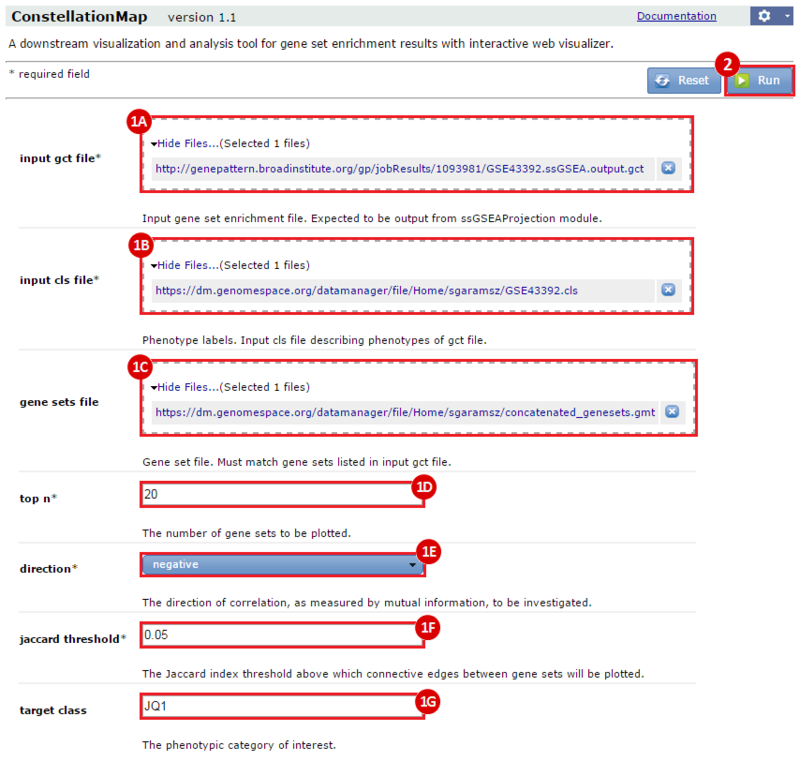
This is an example interpretation of the results from this recipe. First, we identified sets of genes that are significantly up- or down-regulated in MYCN amplified vs. MYCN non-amplified neuroblastoma tumors. We built a personalized collection of gene sets by combining these two new gene sets, MYCN_amplified_up and MYCN_nonamplified_up, with the Hallmarks collection gene set collection from MSigDB.
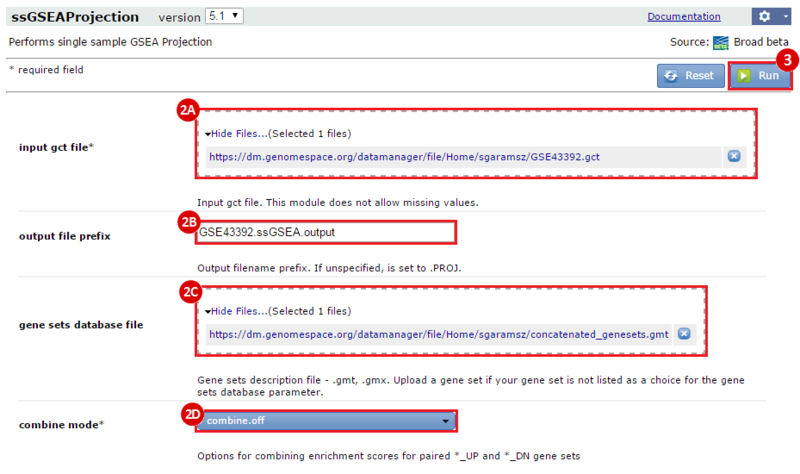
Next, we investigated whether any of the gene sets in our combined collection were significantly enriched in neuroblastoma cell lines treated with DMSO (control) compared with those treated with JQ1. We used the ssGSEAProjection module to get per sample enrichment values for each gene set. Then, we compared these gene set enrichment profiles against each other, and against the JQ1 treatment vs. control phenotype, using the ConstellationMap module.
Given the effects of JQ1, we expect that MYC and MYCN associated gene sets, including our new signature-based gene sets, will have enrichment profiles that are significantly associated with controls compared to JQ1-treated samples.
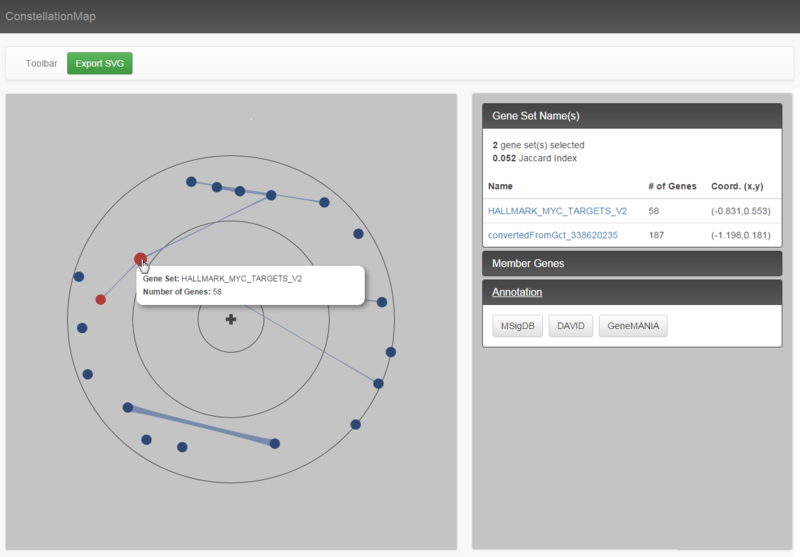
Our results seem to suggest that MYC and MYCN-amplification programs (highlighted) are indeed affected by JQ1 treatment, being highly anti-correlated to JQ1-treated samples and having similar enrichment profiles. Moreover, these gene sets share 12 genes that may be further investigated as markers for JQ1 susceptibility. As investigators, we could continue to investigate clusters of any of the other highly associated gene sets for potential leads in the search for downstream effects of JQ1 treatment. As these are investigative steps, the significance of any results would need further confirmation.

 GenomeSpace.org
GenomeSpace.org