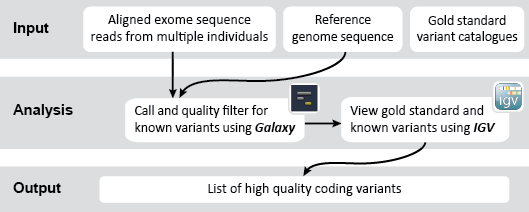
This is an example interpretation of the results from this recipe. In our Galaxy workflow we first used FreeBayes to compare our short-read exome sequencing alignments to a reference sequence in order to determine likely genetic variants in our sample cohort. Next we used VCFfilter to filter these results for high quality variants based on various metrics (e.g., read depth, quality score). We also used VCFintersect to further narrow our results down to those variants with equivalent alleles in the HapMap catalog. Finally, we turned to IGV to view our filtered variants alongside a set of known pathogenic variants identified in the dbSNP database. Specifically, we would like to see if any of our identified variants may be associated with an increased risk for disease.
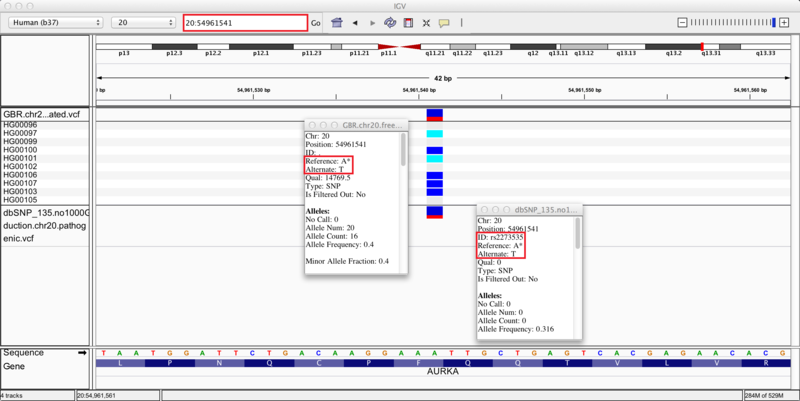
We zoom in on a specific variant located in the q13.31 cytoband of chromosome 20 by typing the coordinates 20:54961541 into the regions navigation bar and hitting enter. Both our results track (upper) and dbSNP track (lower) display a variant at this site, as indicated by the blue and red bar (the proportion of red in the bar corresponds to the allele frequency of the variant). The group of 10 tracks display the genotypes of the 10 individuals whose sequences we used in this recipe. Specifically, 4 individuals are heterozygous for the variant (blue), 2 individuals are homozygous (cyan), and the remaining 4 individuals are homozygous (grey) for the reference allele.
Clicking on the variants in either track brings up info-boxes that display various statistics for the variant. By comparing to the dbSNP track, we notice that our variant analysis has identified an A/T SNP that corresponds to the pathogenic variant, rs2273535, in the AURKA gene. Moreover, our set of 10 samples carried the minor allele with a frequency of 0.4, which corresponds closely to the allele frequency of 0.316 reported in the dbSNP annotations. Given this correspondence, we reason that we have identified the pathogenic SNP rs2273535 in our sample set of 10 British individuals. If we search for SNP id rs2273535 in ClinVar (link), we see that it has been identified as a risk factor for colon cancer. However, note that due to our small sample size these variant discoveries may not be generalizable to the whole population; this is only a simple representation of possible results.

 GenomeSpace.org
GenomeSpace.org

 icon in the upper right corner to import the workflow.
icon in the upper right corner to import the workflow.


Hi, I've been trying to follow this recipe, but could not launch the data to Galaxy. Please check, whether there is any problem with that. Thanks, Marta