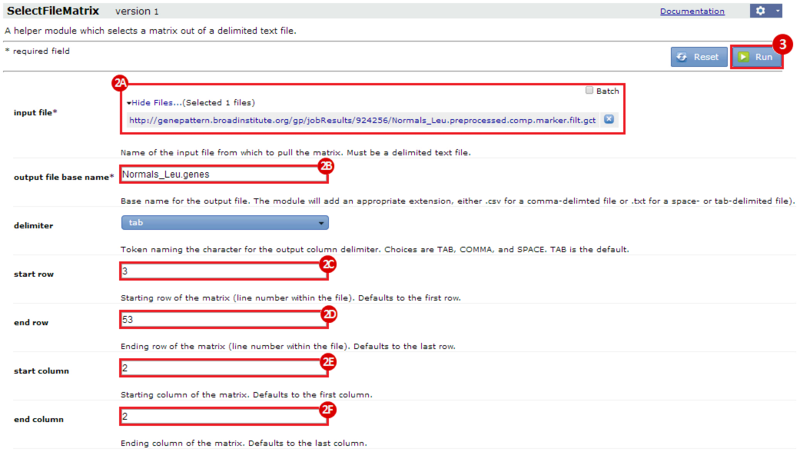
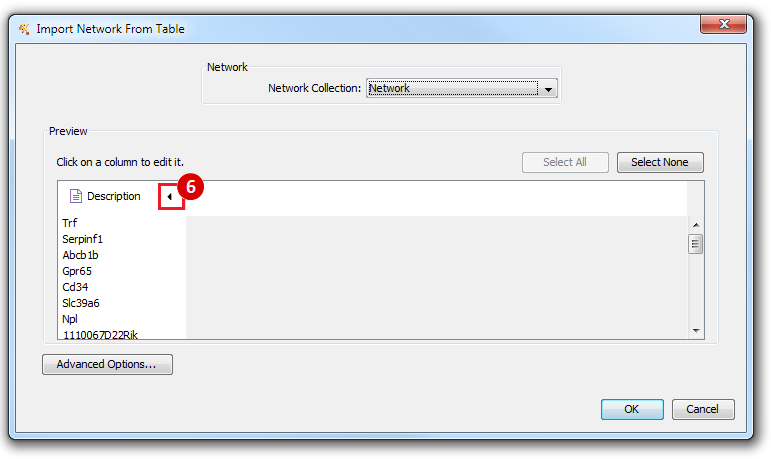
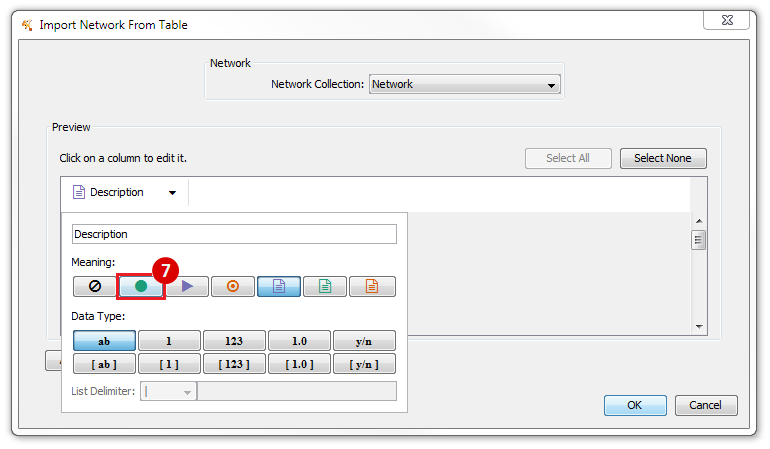
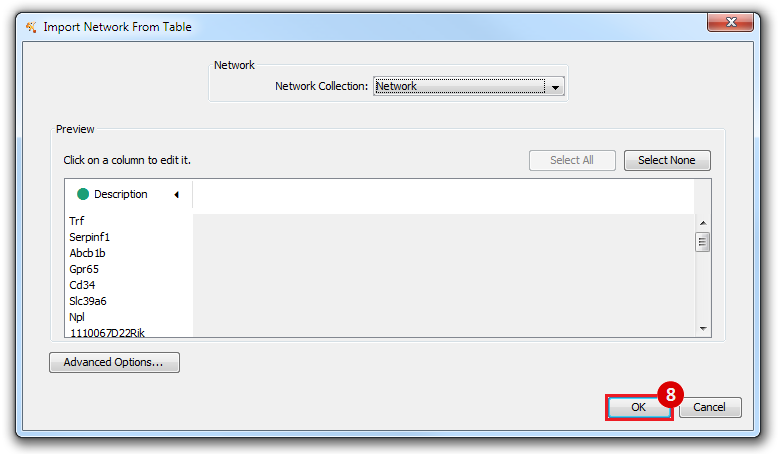
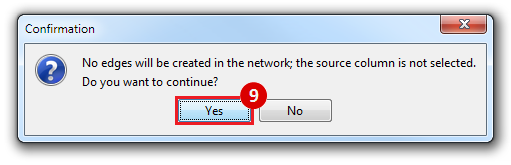
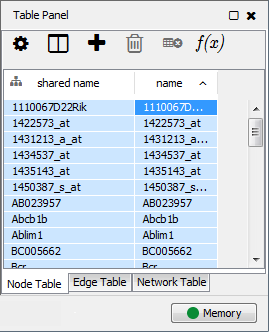
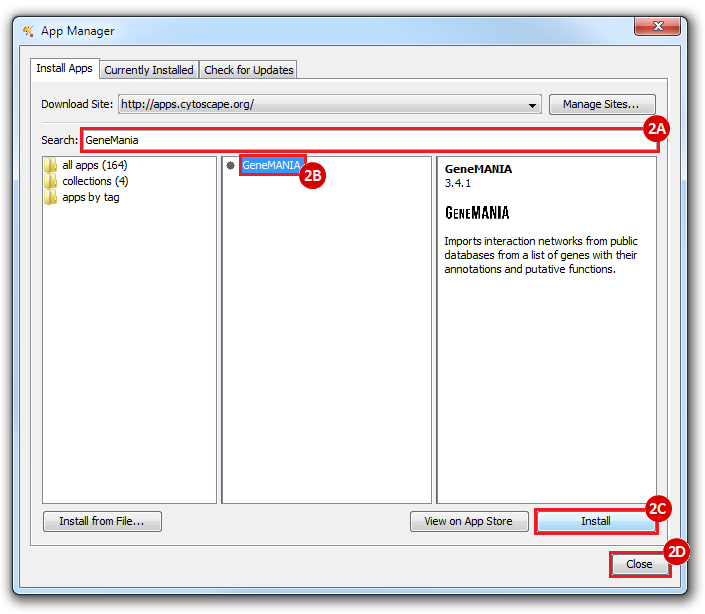
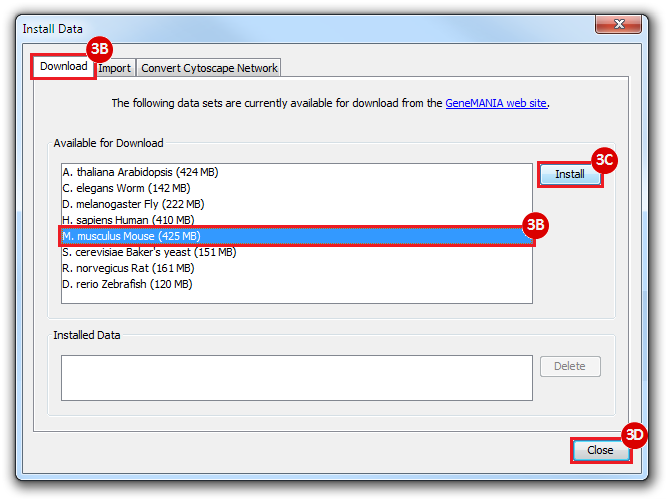
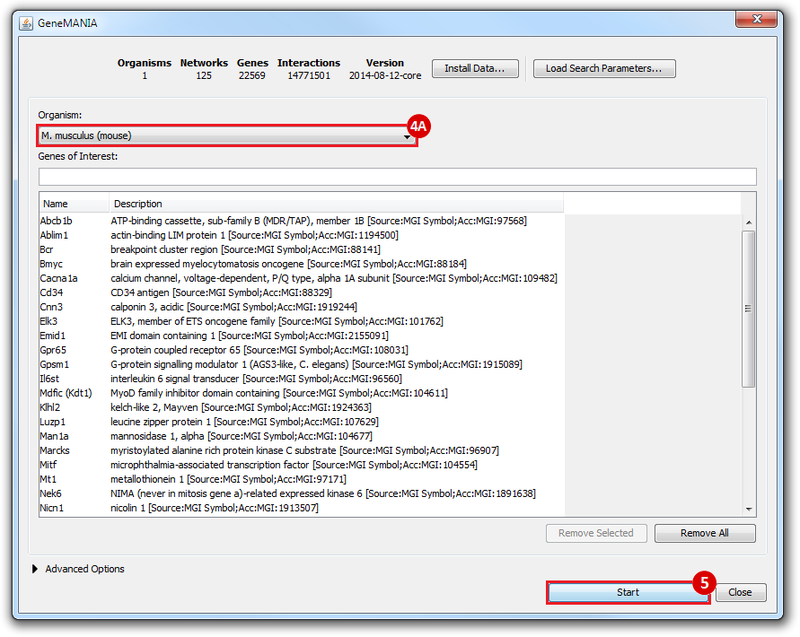
This is an example interpretation of the results from this recipe. First, we identified the top 50 genes which differentiated between two phenotypes, leukemic and normal. We then used the GeneMANIA tool in Cytoscape to identify connections between these genes, although some genes were not annotated and therefore only a subset were actually analyzed. We included all possible sources of interaction, i.e. we are equally interested in connections between genes that arise from co-expression, as we are in connections arising from physical interactions.
After running GeneMANIA we created a network which connected our subset of genes. We can see from the GeneMANIA results that, e.g. 3 genes (out of 45) have the Gene Ontology (GO) annotation 'zinc ion homeostasis', which has a total of 16 genes associated with it. The significance of this enrichment is reported as a q-value, calculated from a FDR corrected hypergeometric test for enrichment. The q-value is analogous to a p-value, and therefore a lower q-value is considered more significant.
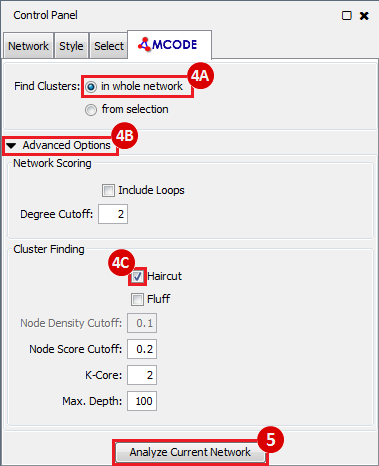
Once we learn about the functional enrichment associated with the genes in our network, we are interested in determining whether we can find subnetworks, i.e. areas of the network which have motifs. We use MCODE to identify subnetworks. For example, MCODE identifies a small subnetwork of 5 nodes and 6 edges. When we click on this subnetwork in the MCODE panel, it will highlight the nodes in the network.
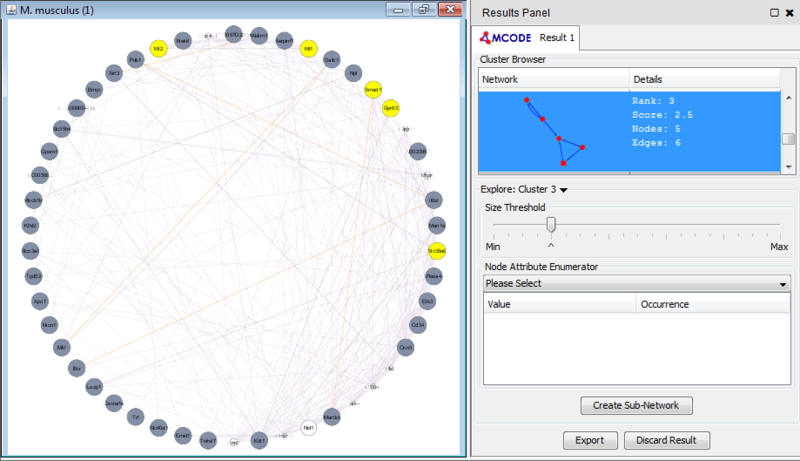
We can separate this set of genes into a subnetwork by clicking the Create sub-network button in MCODE. This generates a new view containing the subnetwork, arranged according to the MCODE subnetwork view. Looking at this subnetwork of genes, we can examine their annotation and see that 3 of these genes are associated with zinc ion binding.
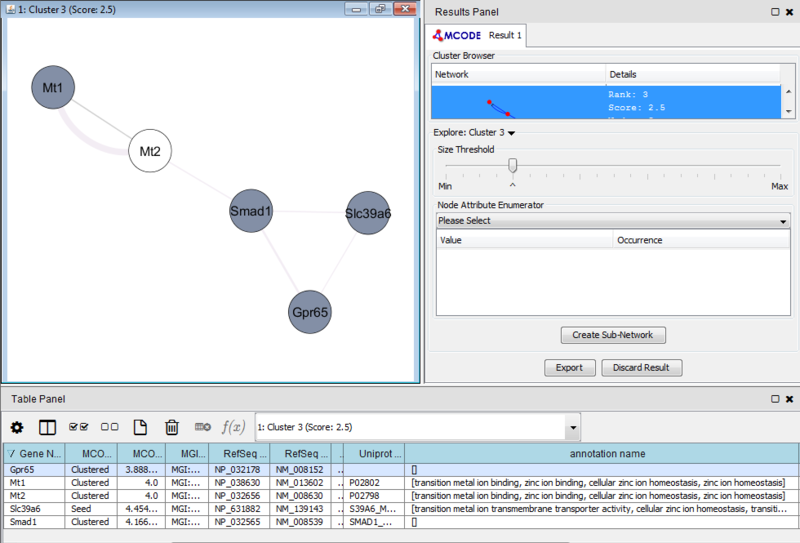
These results suggest that there is a collection of zinc ion binding genes in our set of 50 genes which differentiated between leukemic and normal phenotypes. However, the results in this example are not necessarily significant and are only a simple representation of possible results.

 GenomeSpace.org
GenomeSpace.org