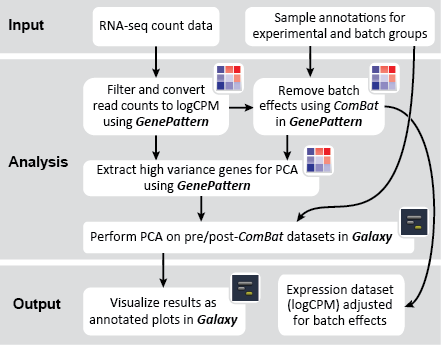
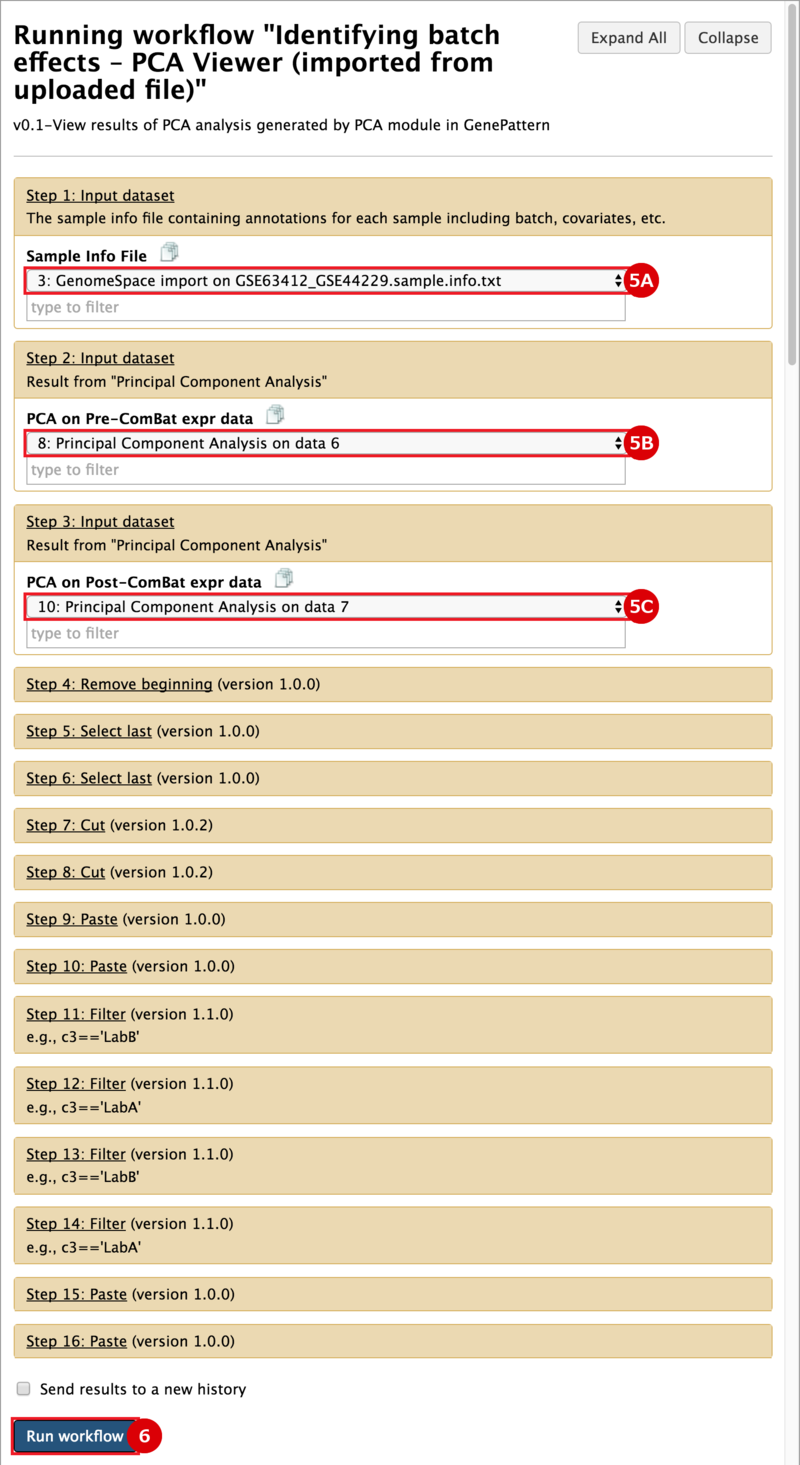
There are two primary goals of this recipe: (1) identify potential batch effects in our dataset and (2) adjust our data for any identified batch effects. This recipe generates a number of plots to help us evaluate our data along these aims:
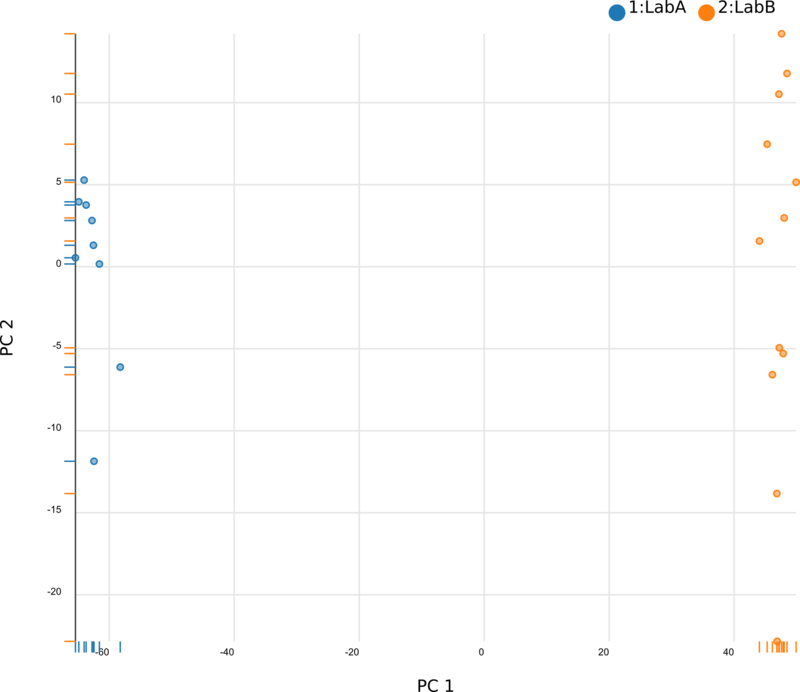
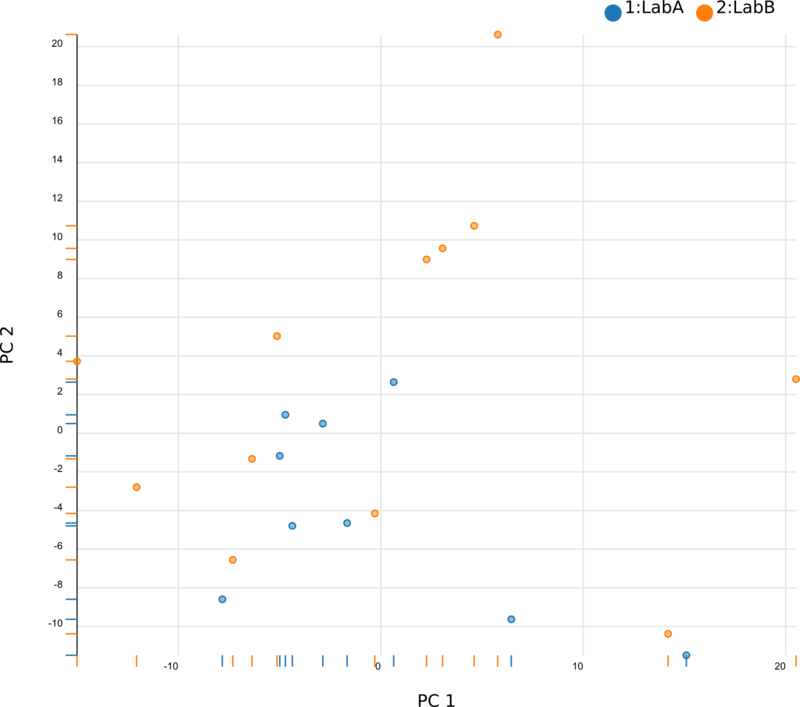
We began by using Principal Component Analysis (PCA) to view the underlying structure of our raw expression data prior to applying ComBat. To do this, we projected our data in the space of the first two principal components, PC1 and PC2, with each point representing a different sample from the experiment (Fig. 1). The PCs identified by PCA transformation are ordered such that the first principal component, PC1, captures the largest possible variance in the dataset, and each following PC captures the highest variance possible while being orthogonal to the previous PCs. If we view our pre-ComBat data, we clearly see that the samples cluster according to the lab (Lab A or Lab B) that they came from. Since these clusters are located at disparate ends of PC1, it appears that the originating lab introduces much (if not most) of the variance in our dataset. We conclude that the lab in which the samples were processed is a batch effect.
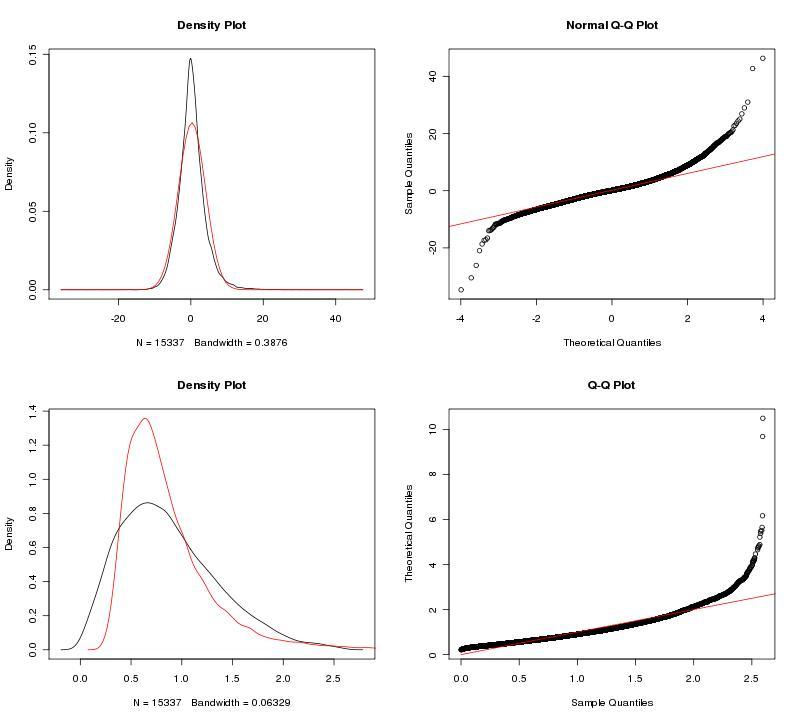
To adjust our dataset for the identified batch covariate, Lab, we used ComBat. ComBat requires that we specify this batch covariate as well as covariates of interest; in this case, we were interested in a single covariate, treatment (control, fear conditioning, object location memory). The parametric ComBat method that we used computes a prior probability distribution for the adjustment. To evaluate this prior distribution, we view the prior plots that the ComBat algorithm outputs (Fig. 2). In this case, the parametric estimate (red) overlaps the "true" distribution (black, kernel density estimate) pretty well, so we can feel reasonably confident in our ComBat adjustment. In the case of poor estimates, we would resort to the non-parametric setting for the algorithm. For more information, see GenePattern's documentation on ComBat.
After applying ComBat to our expression dataset, we again apply PCA and project the transformed data into the space of the first two PCs (Fig. 3). Post-ComBat, we see that the samples from our two lab groups are more overlapping, indicating that the batch effect has been mitigated.
At this point, the investigator can proceed in number of directions. The post-ComBat PCA projection can be explored to see how our covariates of interest cluster. Moreover, the ComBat-adjusted dataset (GSE63412_GSE44229.logcpm.combat.gct) can be used in downstream expression analysis (e.g., differential expression analysis).
References
- Johnson WE, Rabinovic A, and Li C. Adjusting batch effects in microarray expression data using Empirical Bayes methods. Biostatistics. 2007;8(1):118-127. PMID: 16632515
- Ma S and Dai Y. Principal component analysis based methods in bioinformatics studies. Brief Bioinform. 2011 Nov;12(6):714-22. PMID: 21242203
- Peixoto L, Risso D, Poplawski SG, Wimmer ME et al. How data analysis affects power, reproducibility and biological insight of RNA-seq studies in complex datasets. Nucleic Acids Res. 2015 Sep 18;43(16):7664-74. PMID: 26202970
- Vogel-Ciernia A, Matheos DP, Barrett RM, Kramár EA et al. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat Neurosci. 2013 May;16(5):552-61. PMID: 23525042

 GenomeSpace.org
GenomeSpace.org