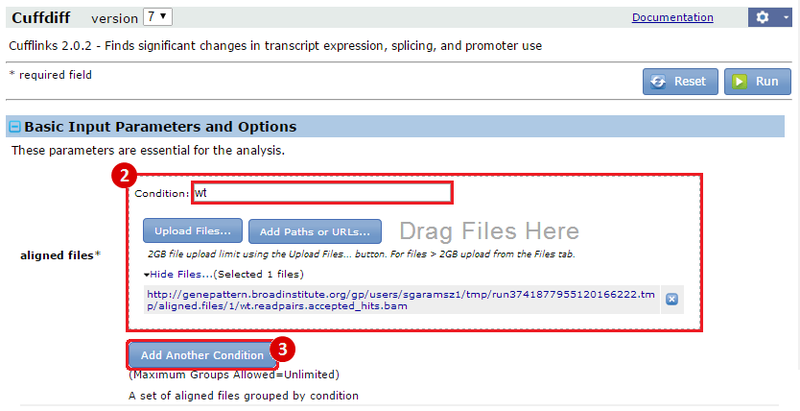
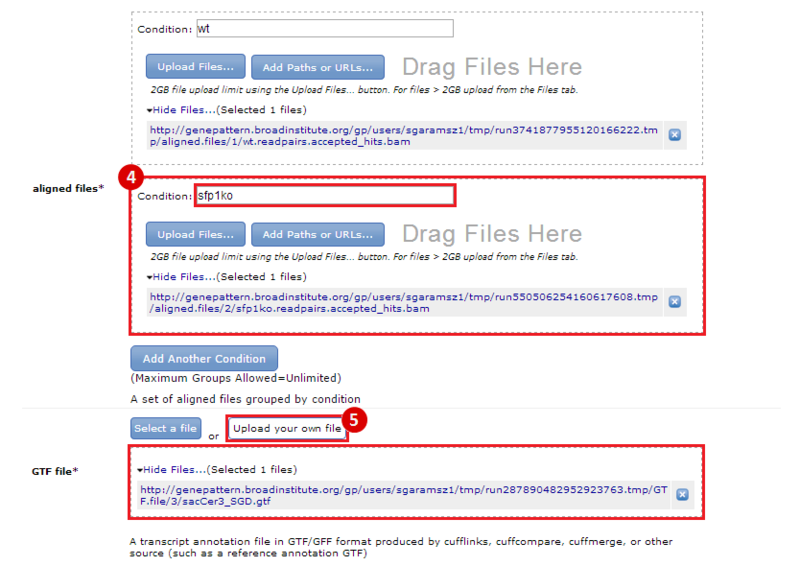
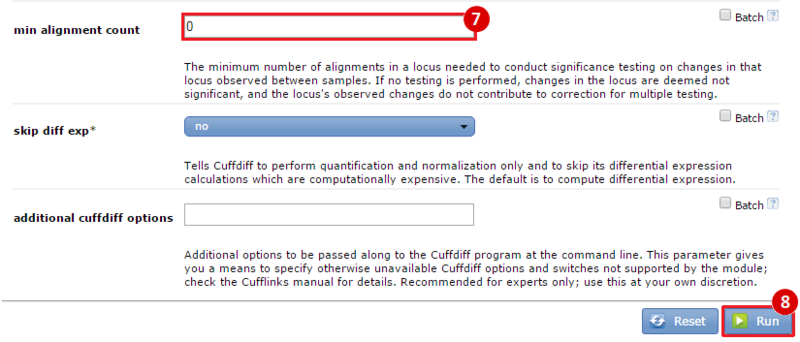
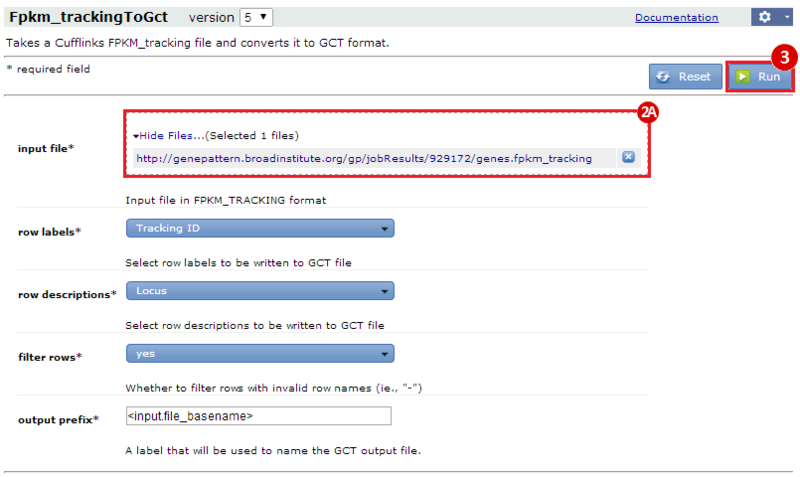
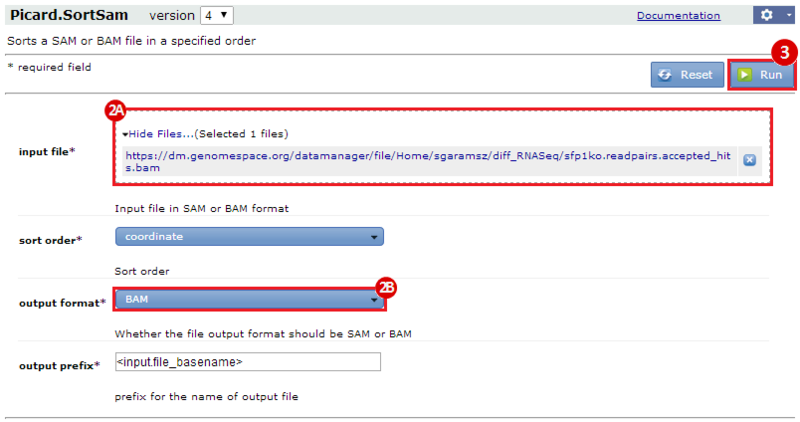
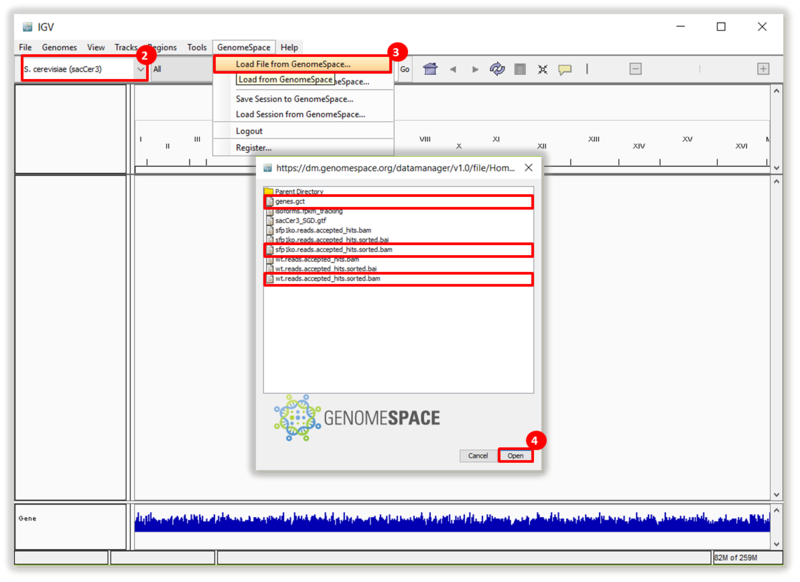
This is an example interpretation of the results from this recipe. First, we used TopHat in GenePattern to align raw reads from RNA-seq to a reference genome (pre-built in Bowtie). After aligning both a wild-type and mutant strain of S. cerevisiae, we used Cufflinks to determine the genes which were differentially expressed when comparing the two phenotypes. We then use IGV to visualize the difference between these two datasets; in particular, we validate that the SFP1KO mutant strain of S. cerevisiae lacks the SFP1 gene.
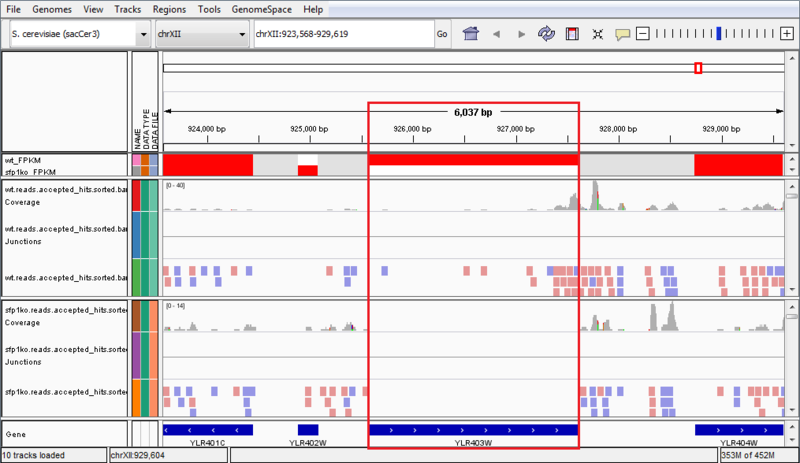
The wt_FPKM track is red in the region of the SFP1 gene, whereas the sfp1ko_FPKM track is white. This is an indicator of gene expression, and shows that the SFP1 gene is not being transcribed in the SFP1KO mutant strain. We can see an additional indicator of this when we compare the wt.reads.accepted_hits.sorted.bam file to the sfp1ko.reads.accepted_hits.sorted.bam file. These are the tracks which have red and blue boxes to indicate where reads aligned against the reference genome. We can see there are several reads aligned in the WT strain to the SFP1 gene, but the area is blank in the SPF1KO mutant, showing that no reads from that gene aligned against the reference genome. These results confirm that the SFP1 gene was knocked-out in the SFP1KO mutant strain of S. cerevisiae. However, the results in this example are not necessarily significant and are only a simple representation of possible results.

 GenomeSpace.org
GenomeSpace.org



 next to the file in the
next to the file in the