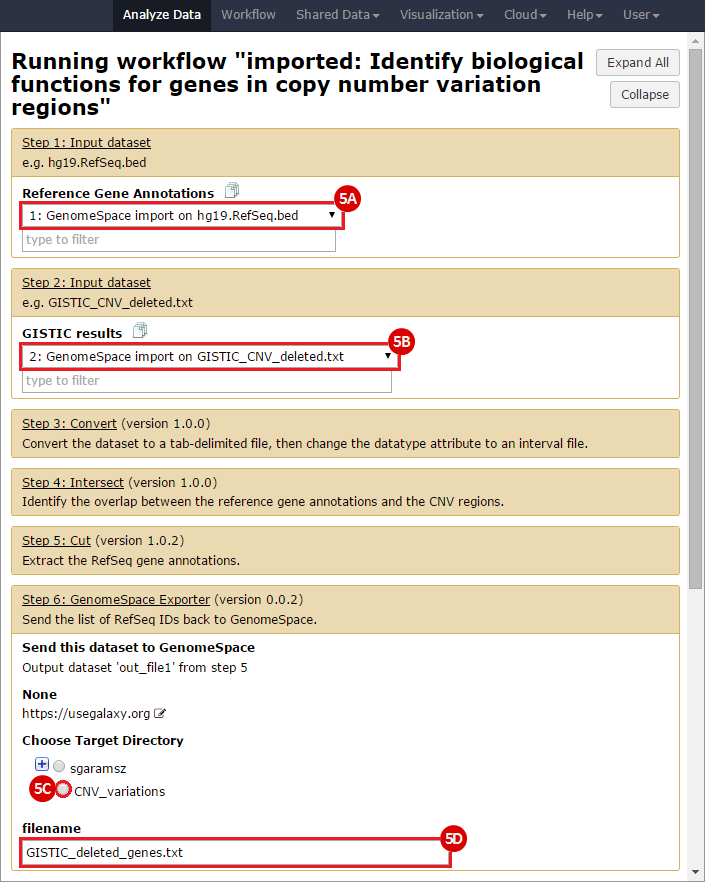
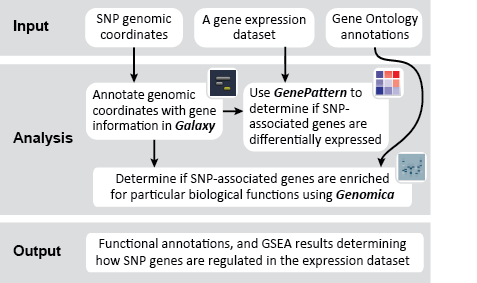
This is an example interpretation of the results from this Recipe. First, we identified the overlap between reference gene annotations (RefSeq format) and the copy number variation (CNV) regions using Galaxy. This results in a list of annotated genes that are located in the CNVs; there may be more genes in the CNV regions that are not properly annotated and therefore were missed in the analysis. In this example, we find roughly 1300 genes that are amplified in CNV regions, and roughly 8800 genes that are deleted in CNV regions. Next, we were interested in knowing what, if any, functional annotation these genes had - are there specific gene functions being duplicated in CNV regions? Are the gene products in these regions connected functionally?
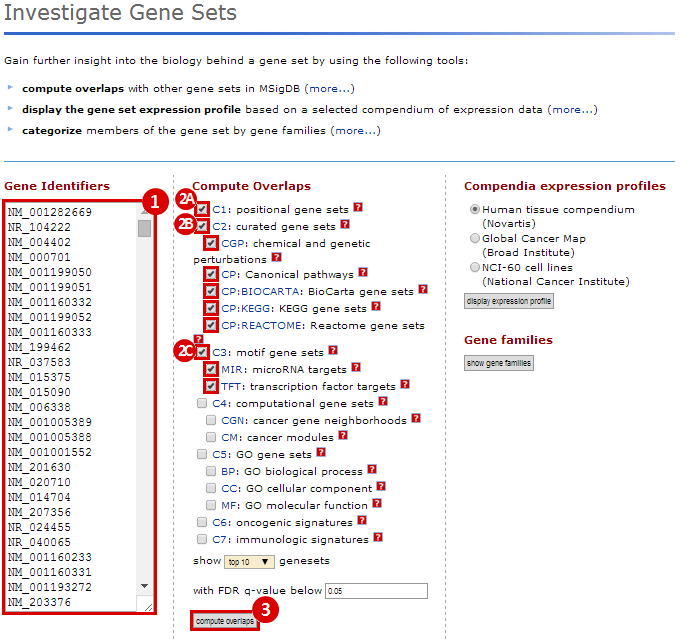
We used MSigDB to probe our dataset for functional annotation. In this case, we used only three collections: C1, C2 and C3. In this example we are most interested in knowing whether our genes are related to chromosomal deletions or amplifications (C1: positional gene set), whether our genes have functions that are reviewed in the literature (C2: curated gene set), and whether our genes share any cis-regulatory motifs (C3: motif gene set).
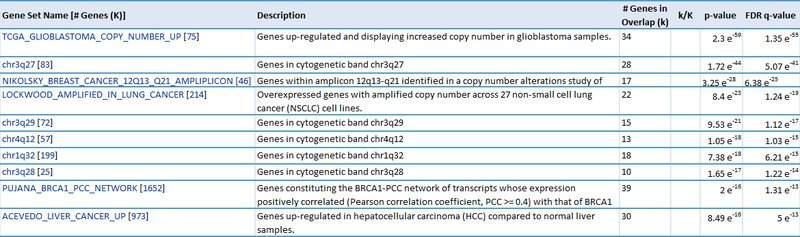
Our first result lists the gene set name and description, the number of our genes which overlap with the gene set, and measures of significance (p-values and q-values). For example, we see that 28 genes out of the ~1300 amplified genes fall into the "chr3q27" category, which has 83 genes total. This result is significant (p-value = 1.72e-44). This suggests that the CNV regions associated with glioblastoma are enriched for genes duplicated on chromosomal region chr3q27. Similarly, 222 genes out of the ~8800 deleted genes fall into the "chr19q13" category, suggesting that glioblastoma is associated with deletions in this chromosomal region (p = 3.78e-135). This is just one example of a possible interpretation of these results.

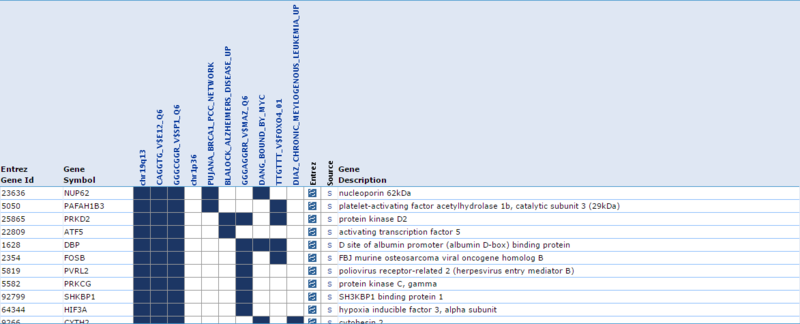
Our second result lists each gene by ID and Symbol, then highlights which of the top categories it is in. For example, the amplified gene CDK4 overlaps with 4 categories: TCGA_GLIOBLASTOMA_COPY_NUMBER_UP, NIKOLSKY_BREAST_CANCER_12Q13_Q21_AMPLICON, LOCKWOOD_AMPLIFIED_IN_LUNG_CANCER, and PUJANA_BRCA1_PCC_NETWORK. If we examine the categories, they suggest that CDK4 is amplified in glioblastoma, breast cancer, and lung cancer, and that CDK4 is a part of the BRCA1 regulatory network. Similarly, when we examine the deleted gene NUP62, we observe that it overlaps with 5 categories: chr19q13, CAGGTG_V$E12_Q6, GGGCGGR_V$SP1_Q6, PUJANA_BRCA1_PCC_NETWORK, and DANG_BOUND_BY_MYC. This suggests that NUP62 is in the chr19q13 chromosomal region, that it contains motifs CAGGTG and GGGCGGR, and that it is associated with the BRCA regulatory network and its promoter is bound by Myc.

These results suggest that our gene list is enriched for specific chromosomal regions and specific promoter region motifs, among other functional annotations. This suggests that functionally related genes are being duplicated in CNV regions in a cancer phenotype. However, the results in this example are not necessarily significant and are only a simple representation of possible results.

 GenomeSpace.org
GenomeSpace.org


 icon in the upper right corner to import the workflow.
icon in the upper right corner to import the workflow.