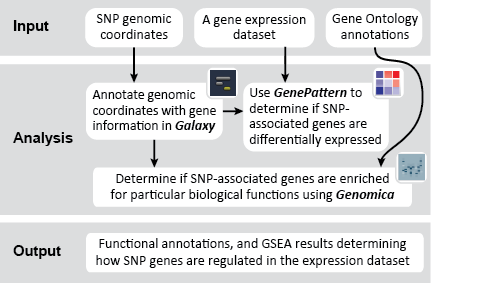
This is an example interpretation of the results from this recipe. First, we performed Gene Set Enrichment Analysis (GSEA) to determine if the SNP-associated genes are up- or down-regulated in a biological condition such as cancer. To interpret our GSEA results, we navigate to the un-zipped file of GSEA results.
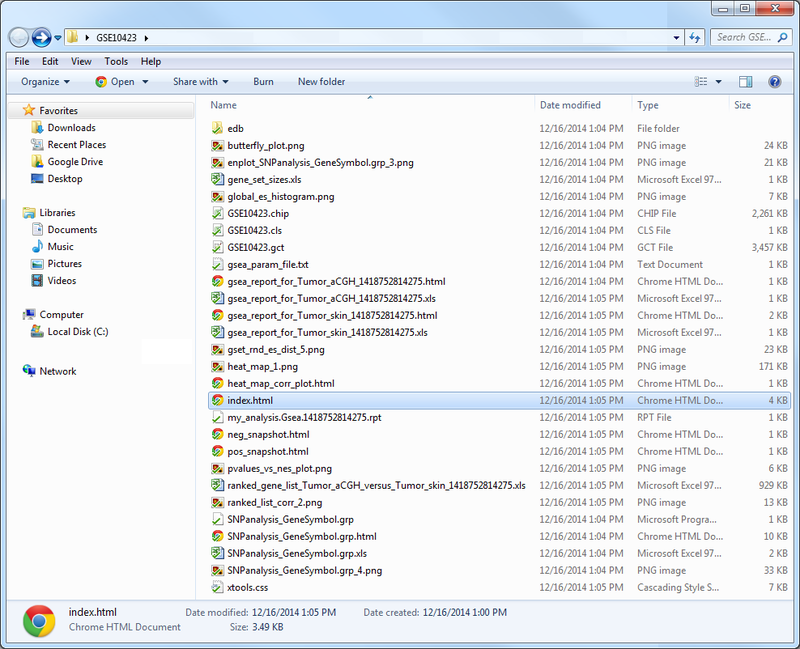
This folder contains the results of the GSEA, including: tables of results; plots such as enrichment plots, heatmaps, butterfly plots, distribution plots, and correlation profiles; and HTML pages designed to display and explain the accompanying results. Clicking on the index.html file will bring up a local HTML webpage linking to the remaining results, in an easy-to-navigate format. In this example, the results indicate that our SNP-associated gene set was up-regulated in the skin tumor phenotype, but these results are not significant. For more help interpreting the results from GSEA analyses, visit the GSEA User Guide page for interpreting GSEA results.
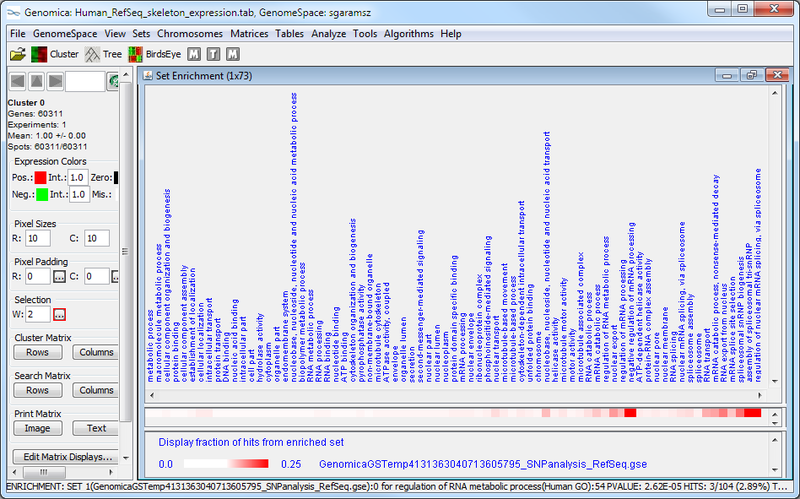
Next, we used Genomica to determine whether SNP-associated genes are enriched for particular biological functions using multiple gene set analysis. The results from this analysis are (1) a table of significantly enriched GO terms, and (2) a graphical representation of the enrichment for each GO term in the gene set being analyzed.
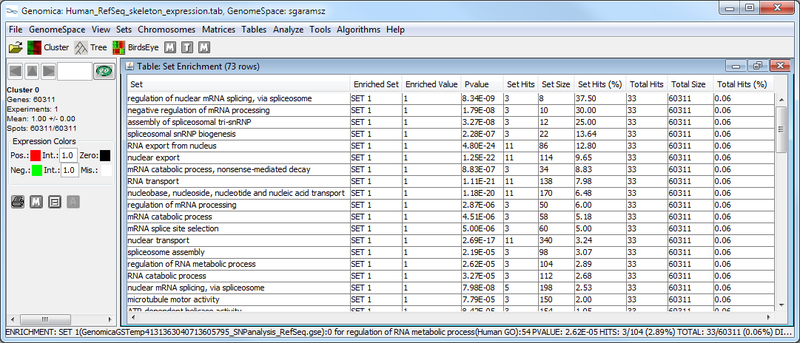
The Genomica results suggest that there is significant overlap between the SNP-associated gene set and GO terms like, e.g., "regulation of nuclear mRNA splicing, via spliceosome" and "negative regulation of mRNA processing". These GO term enrichment results have high p-values (see table above, column 4), and have a large amount of overlap between the SNP-associate gene set and the GO term gene set (see graphic below). The significance of this possible result would need further confirmation.

 GenomeSpace.org
GenomeSpace.org

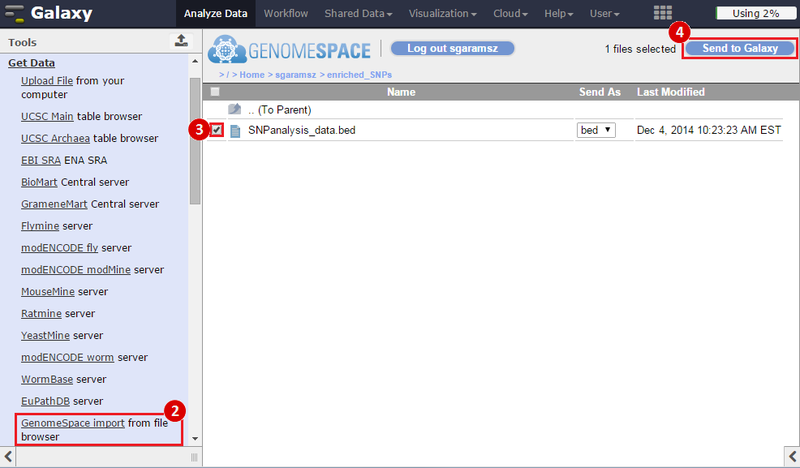
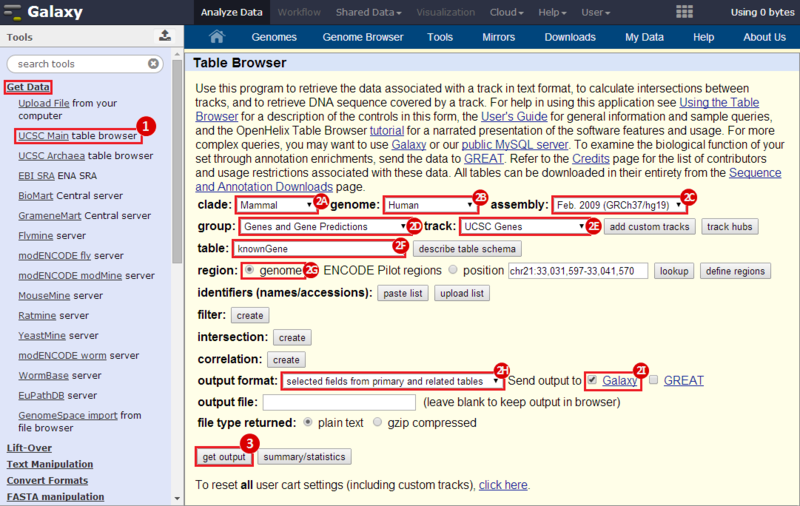
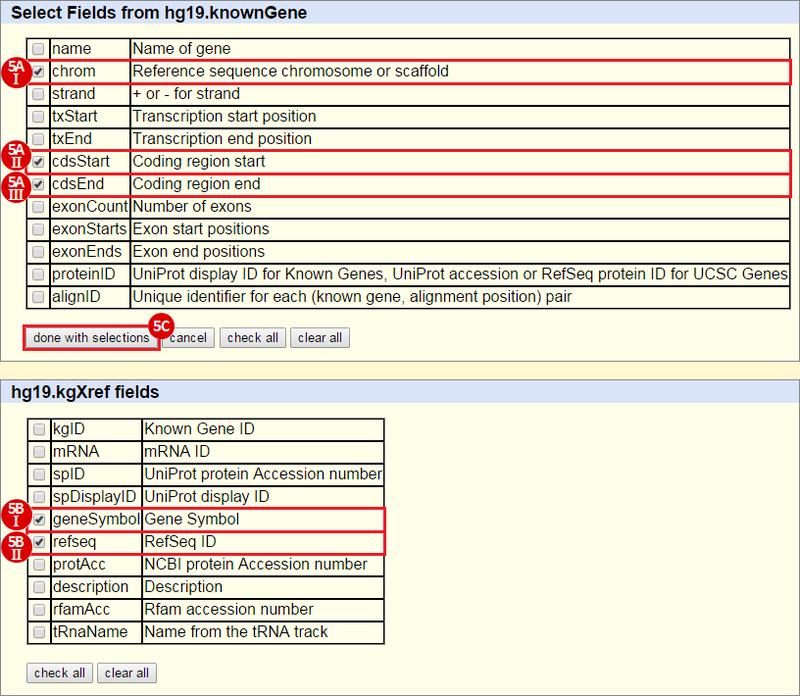
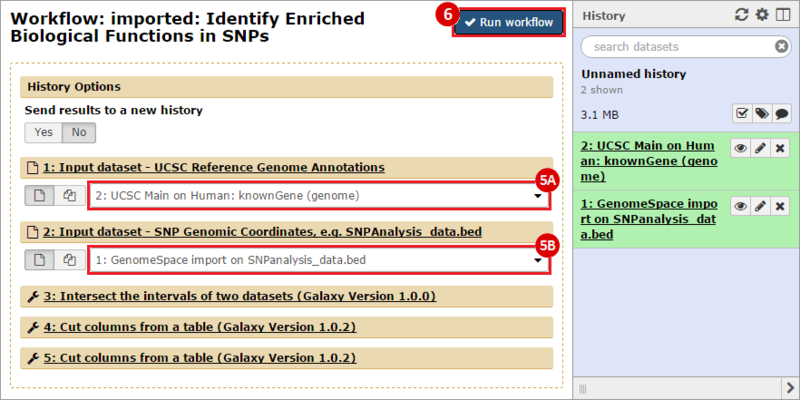
 icon in the upper right corner to import the workflow.
icon in the upper right corner to import the workflow.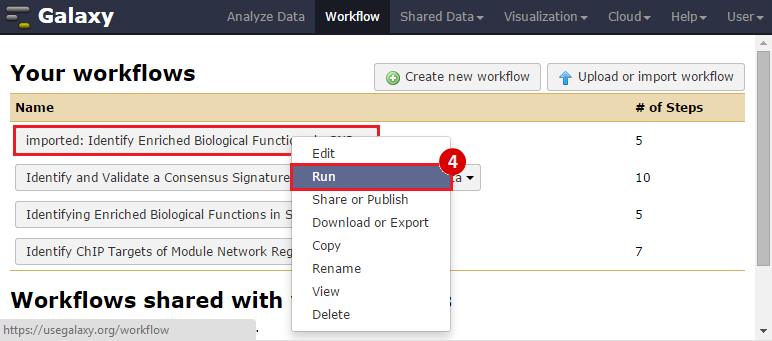
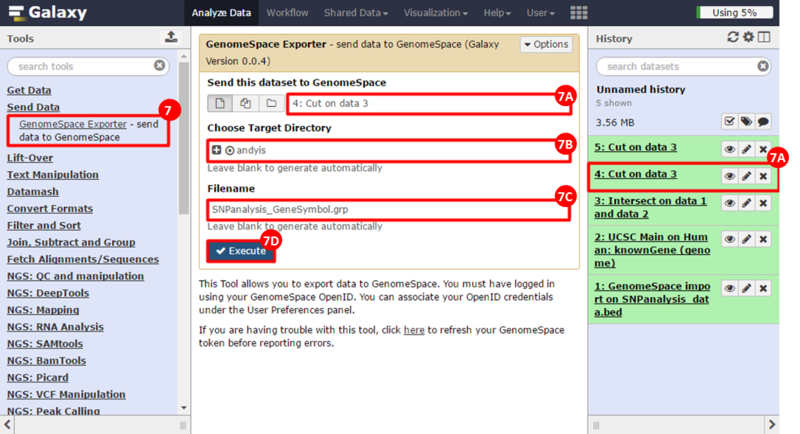

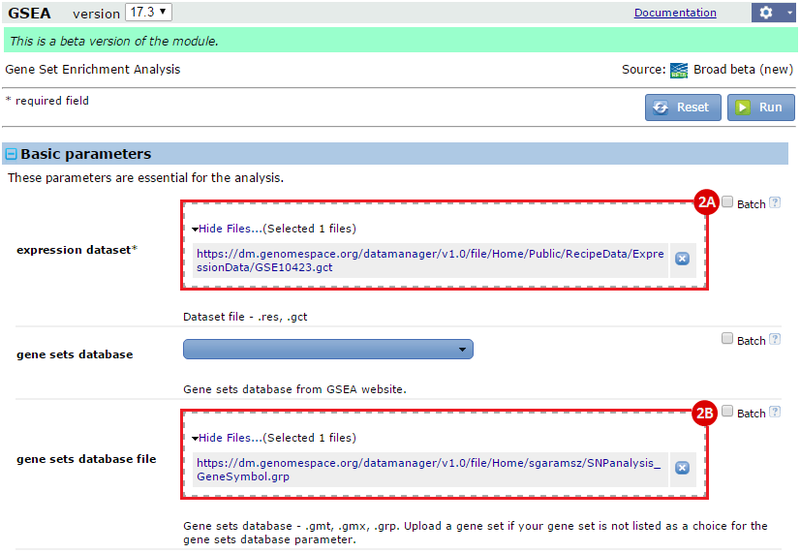
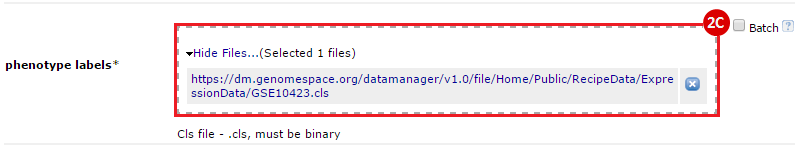
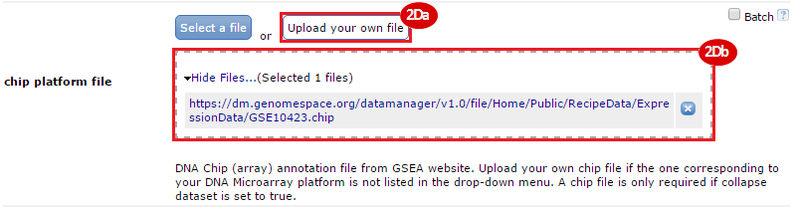
 button to run GSEA.
button to run GSEA.