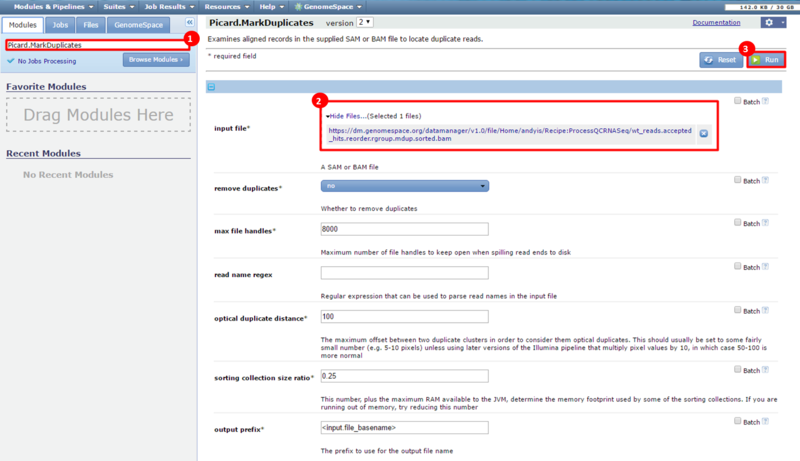
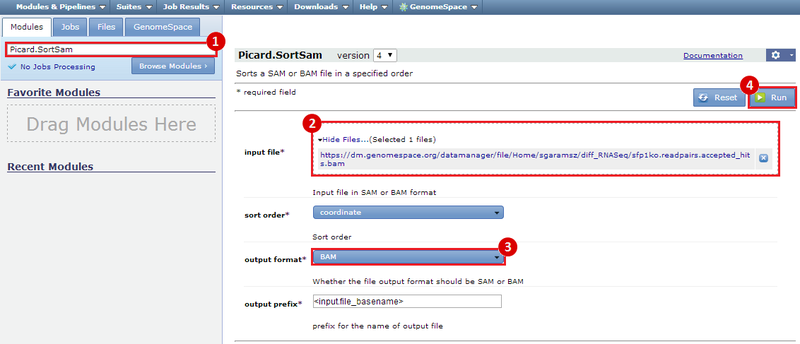
This is an example interpretation of the results from this recipe. In particular, we will focus on interpreting the results from the FastQC tool in Galaxy.
The Per base sequence quality graph shows the quality of each base from the start to the end of a read. In general, areas in green are considered good scores (>30). We can see that toward the end of our reads we have some bad scores, e.g. at position 38, our reads have large error bars which potentially reflect bad scores (>20). The Per sequence GC content graph shows the distribution of GC content for our reads. The blue line indicates the predicted, normal distribution, and the red line indicates the distribution from our read. There is an obvious spike around 55-59% GC content; this spike can indicate contamination from adapter sequences. The Overrepresented sequences table shows that there is one sequence which is observed in over 8% of the reads in our dataset. In addition, FastQC checks the sequences against a list of known adapter sequences, and in this case our sequence matches an Illumina paired end PCR primer. Therefore, we know this is a definite source of error in our reads.
After removing the adapter sequences, we can run FastQC again to see that our data is much improved. The Per base sequence quality graph shows that all the bases in the read have high quality, across the read. In particular, no positions in the read have a quality score which <20 on the y-axis. The Per sequence GC content graph shows that the distribution of GC content in our dataset (red line) is now comparable to the predicted distribution (blue line). This suggests the cause of the GC spike in the original dataset is due to adapter sequences, which we have now removed. Finally, the Overrepresented sequences table shows that only 1.4% of our reads have an adapter sequence. This is a significant improvement, compared to the 8% we had previously.
These results suggest that our RNA-seq dataset was contaminated with adapter sequences and was of poor quality, compared with the dataset after adapter sequences were removed. However, the results in this example are not necessarily significant and are only a simple representation of possible results.

 GenomeSpace.org
GenomeSpace.org

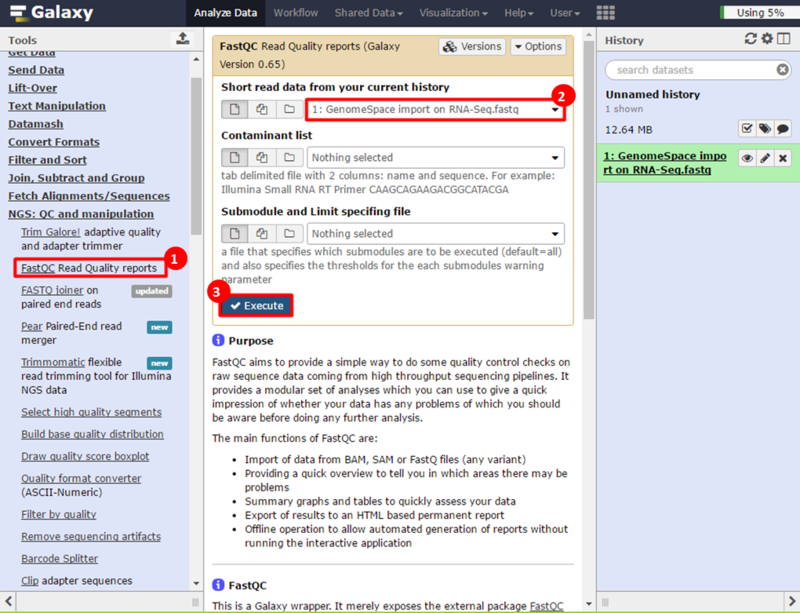

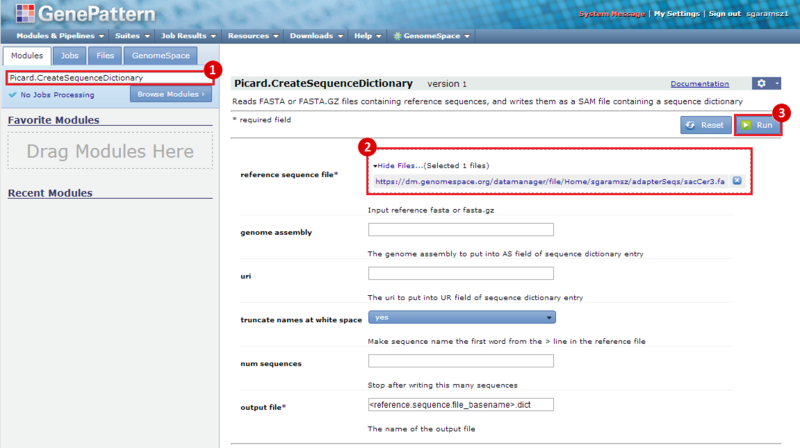
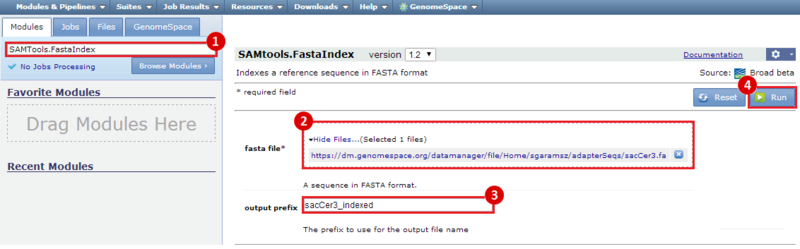
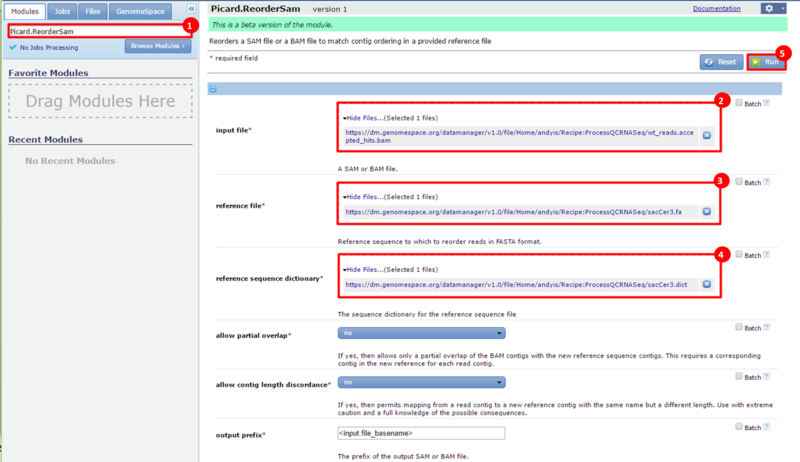
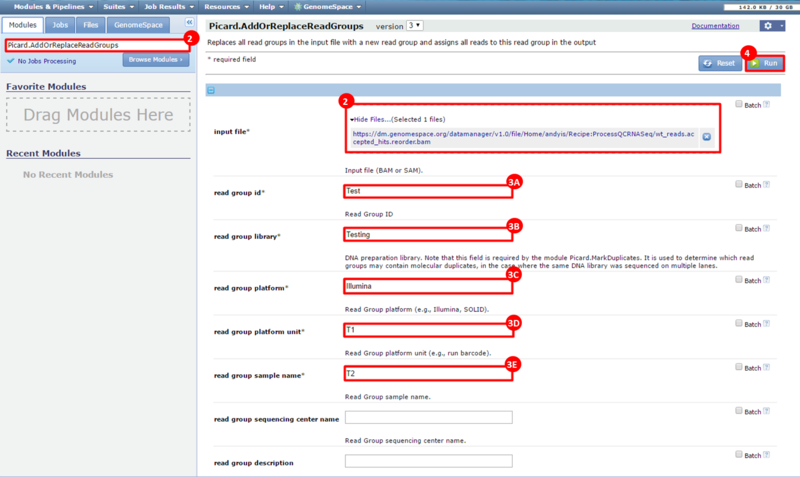
 next to the file in the
next to the file in the