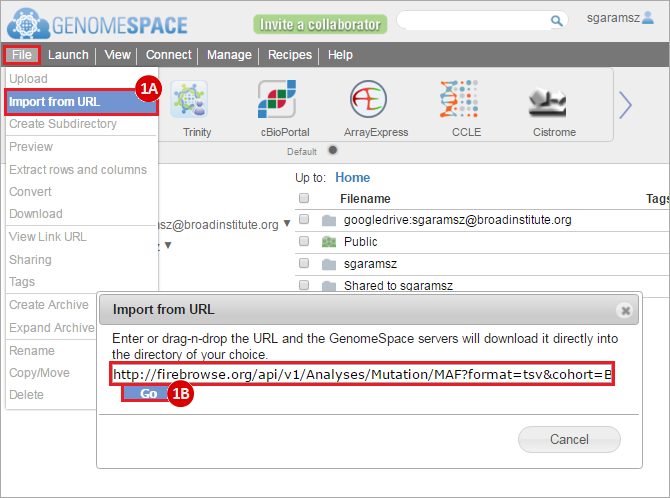
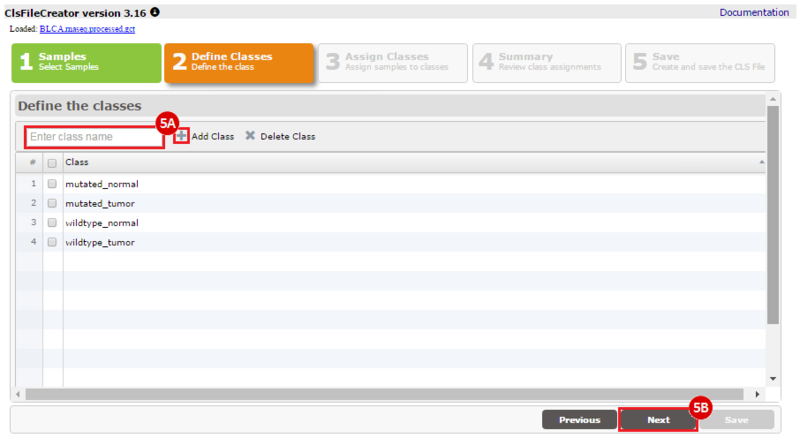
How do I obtain and analyze data from The Cancer Genome Atlas (TCGA)? Which TCGA datasets have specific mutations in my gene of interest?
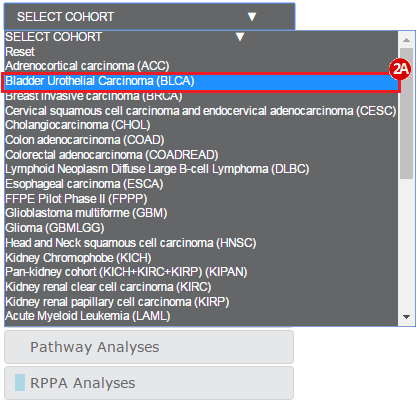
This recipe provides a method for identifying and obtaining specific datasets of interest from The Cancer Genome Atlas (TCGA), through a web-based tool called FireBrowse. An example use of this recipe is a case where an investigator may have a gene they are interested in, such as ERCC2, and would like to know if there are mutations in this gene in specific datasets of interest, such as bladder cancer.
Tumors arise from mutational changes to healthy cells, and are frequently deficient in one or more DNA repair pathways. The accumulation of mutations in tumor can be described by the “mutational signature”, a pattern of genetic mutations found in tumor DNA, which reflect different mutation events. Mutational signatures can be specific to certain tissues or cancer types. Many of these mutational signatures are associated with DNA repair pathways.
An in-depth study of urothelial carcinoma, which causes ~150,000 deaths annually, by Kim et al. (Nature Genetics, 2016) has identified a mutational signature in bladder cancer involving the nucleotide-excision repair (NER) pathway. Kim et al. identified a mutational signature involving ERCC2, a gene encoding a DNA helicase which plays a critical role in the NER pathway. Somatic mutations in this gene may prevent proper functioning of the NER pathway, allowing mutations to accumulate. Uniquely, urothelial cancer is the only known tumor type to date in which ERCC2 is significantly mutated.
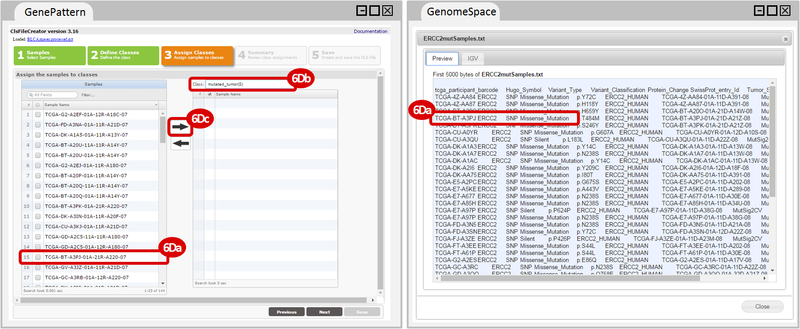
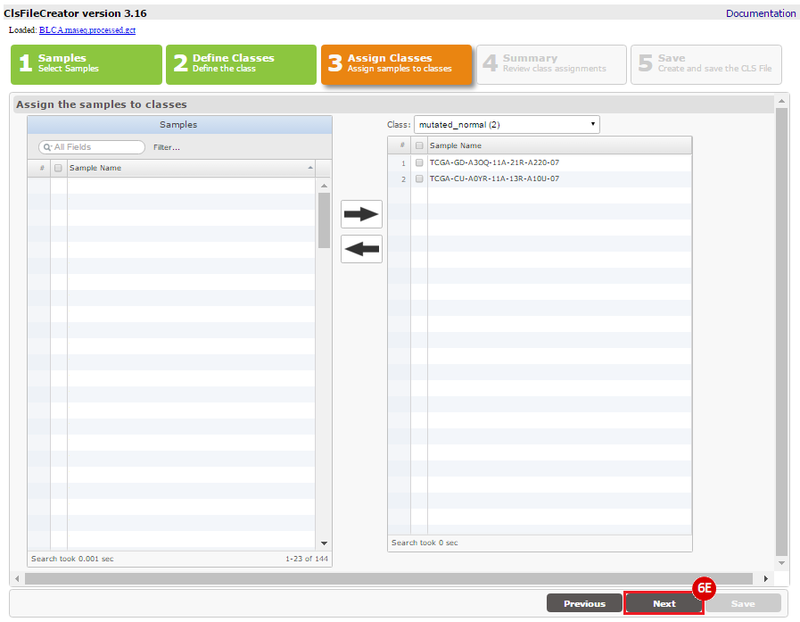
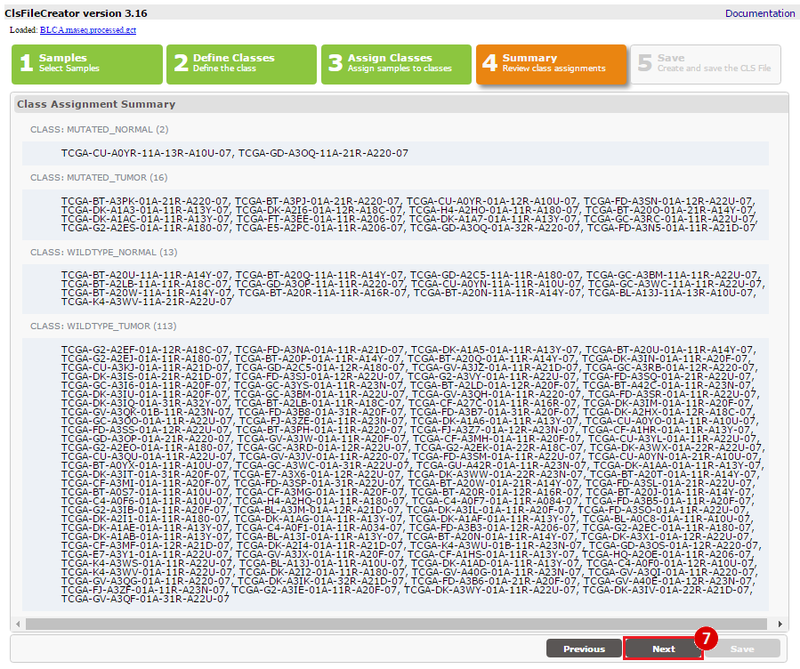
Kim et al. used the collection of bladder carcinoma (BLCA) samples in The Cancer Genome Atlas (TCGA) to complete their analysis. Data were downloaded from the Broad Institute TCGA Genome Data Analysis Center, and samples were categorized based on mutational status. Tumors with somatic, missense mutations in ERCC2 were compared to non-mutated (wild-type) samples to identify the comprehensive mutational landscape of bladder cancer (also described in this TCGA paper).
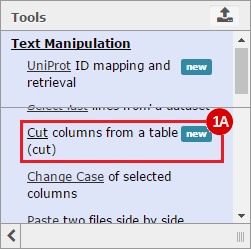
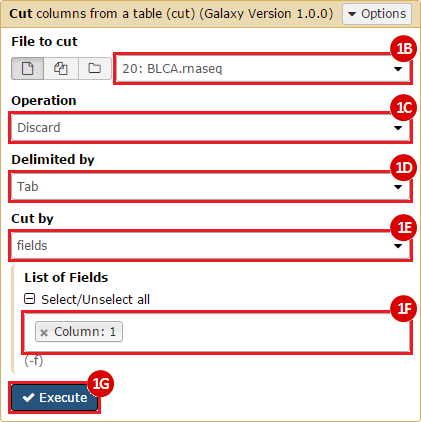
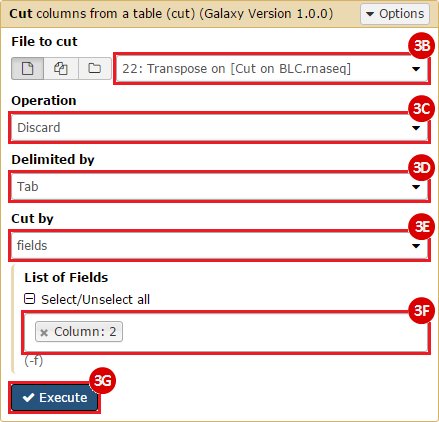
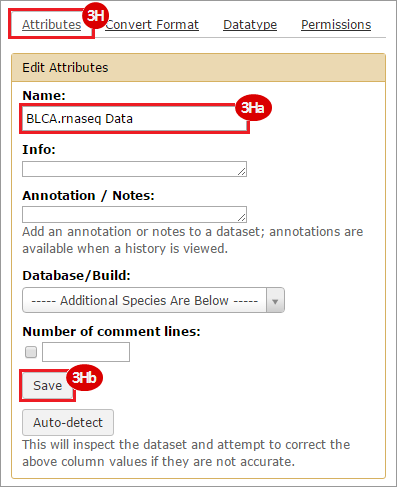
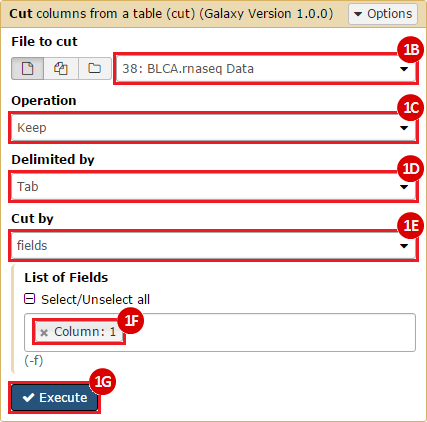
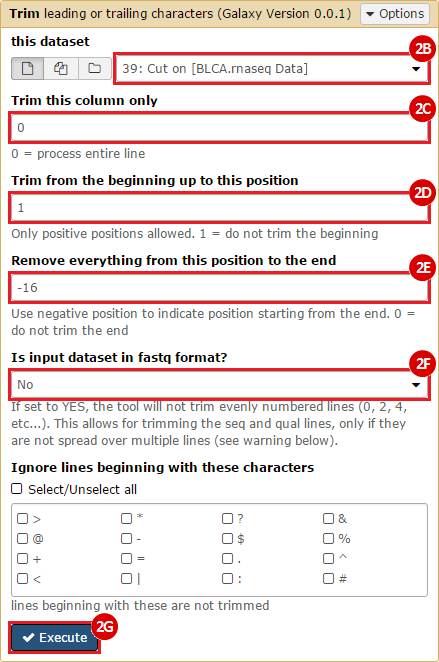
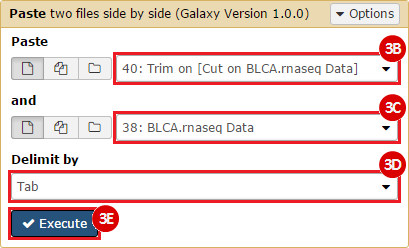
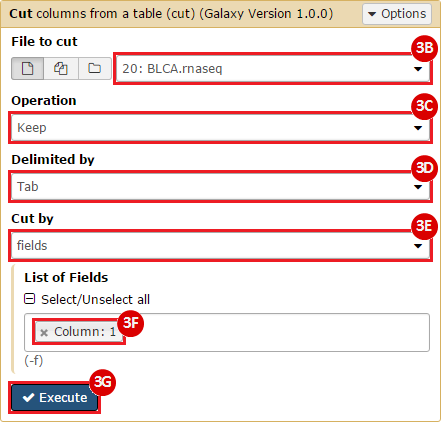
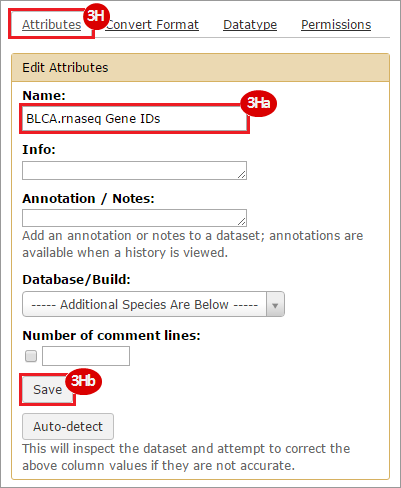
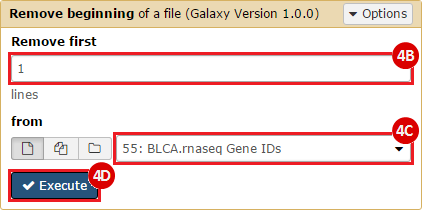
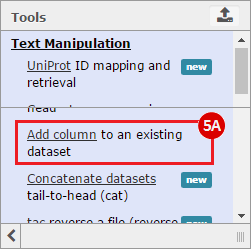
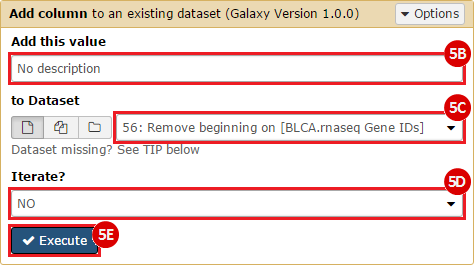
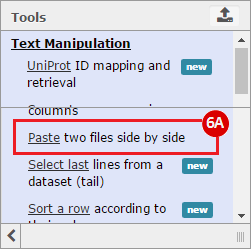
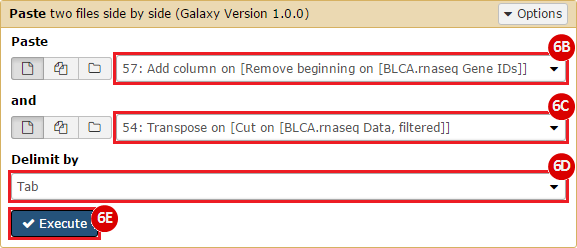
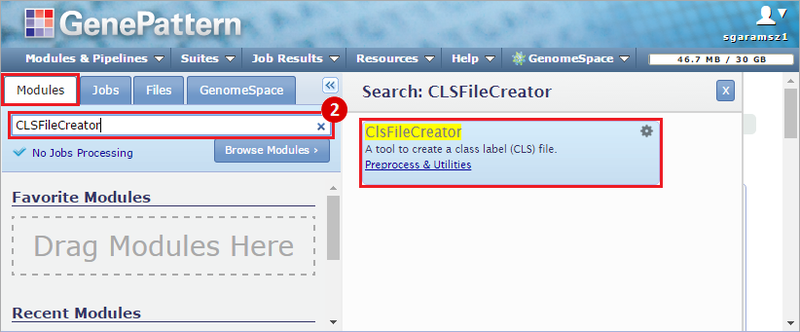
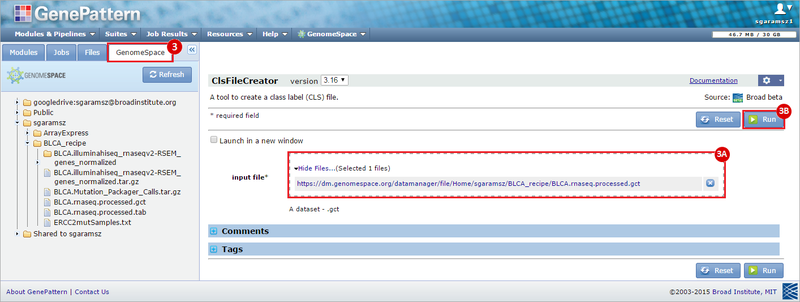
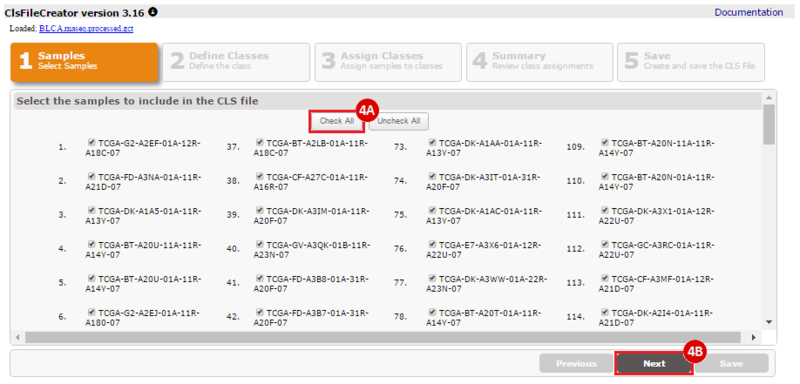
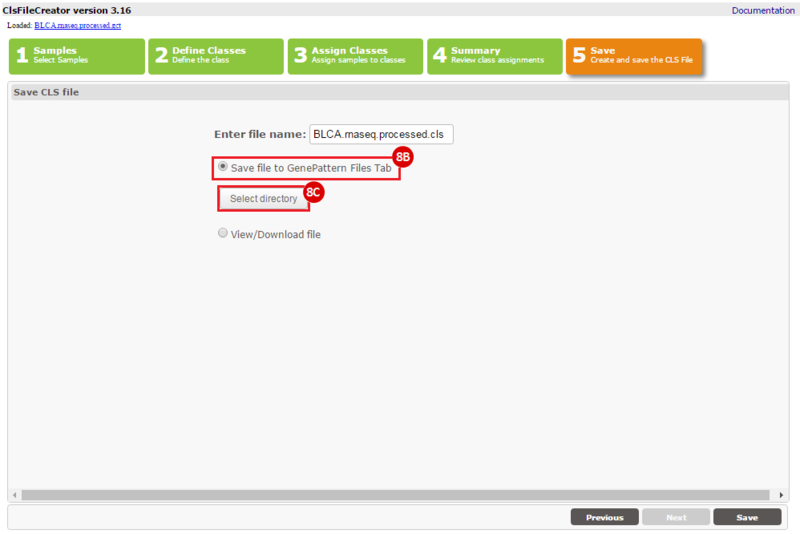
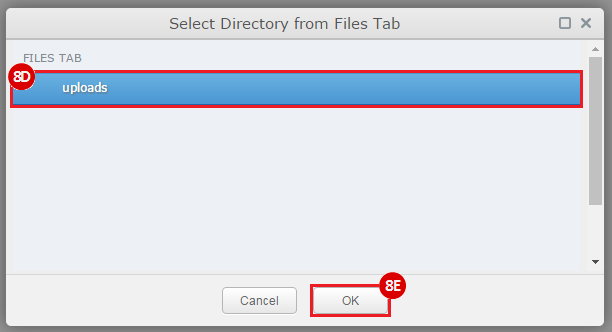
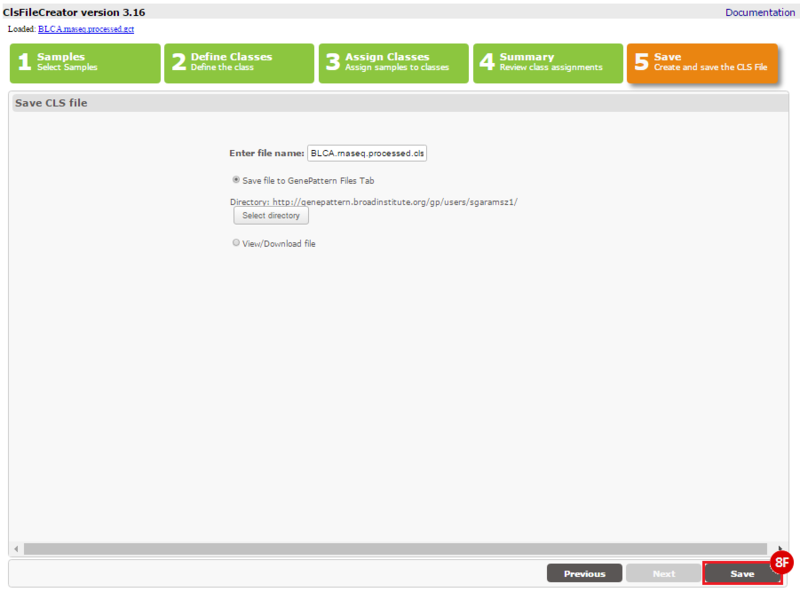
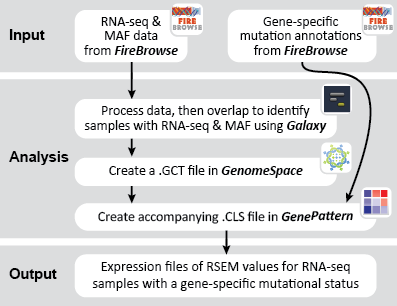
This recipe provides a method for processing data from The Cancer Genome Atlas (TCGA), to identify samples which have mutations in specific genes. The purpose of this recipe is to categorize data by mutational status, for further downstream analysis (e.g. comparing tumors of different mutational status, etc.). Data is collected from FireBrowse; Galaxy and GenePattern are used to categorize samples by mutational status and generate GCT and CLS files. The RNA-seq datasets are gene-level normalized RSEM expression estimates.
TCGA Barcodes
The Cancer Genome Atlas labels its datasets with the TCGA barcode, an identifer that describes the metadata associated with sample. You can learn more about the TCGA Barcodes on the NIH National Cancer Institute Wiki page (see also: working with TCGA data).
TCGA barcodes adhere to a certain format: TCGA-00-1111-22A-33B-4444-55. For this recipe, we are interested in the Sample type, indicated by the 22A section of the barcode. For this recipe we are interested in samples with designation 01 (solid tumor, or TP) or 11 (solid tissue normal, or NT), which are paired tumor-normal samples.
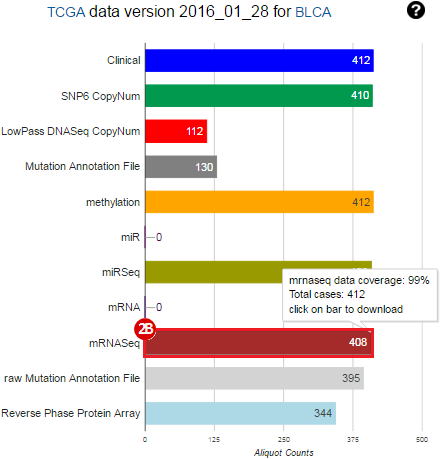
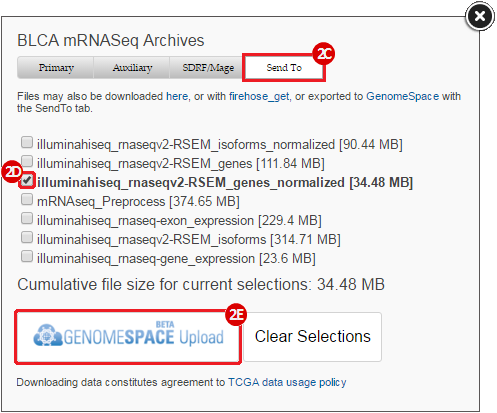
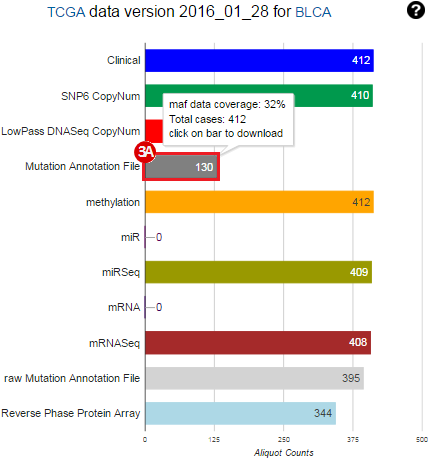
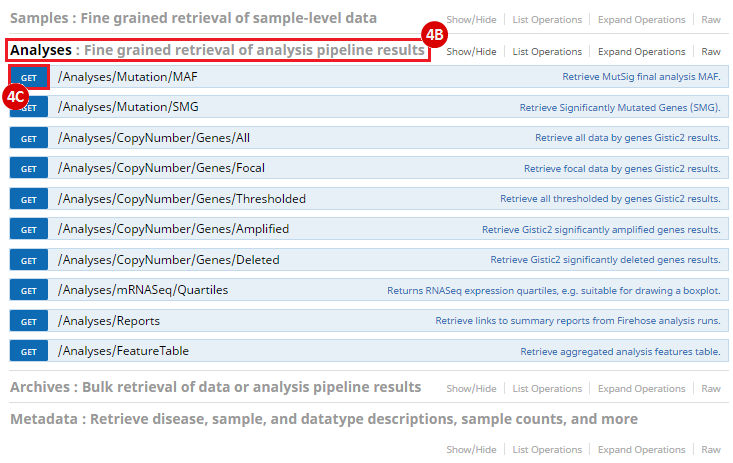
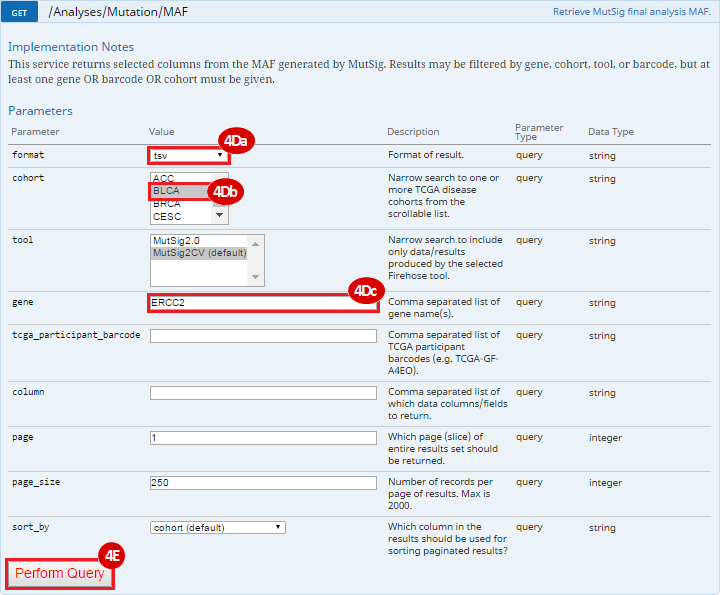
To complete this recipe, we will need several datasets. First, we need example Bladder Cancer (BLCA) data from RNA-seq reads which have been processed and had normalized RSEM values calculated for each gene. In this dataset, we should have bladder cancer tumor samples, in addition to paired normal tissue. For this recipe we are trying to identify the bladder cancer samples that have mutations in the gene ERCC2. Therefore, we also need a list of which samples in the RNA-seq data have mutation annotation files (MAFs), and additionally we need to identify the list of bladder cancer samples that have ERCC2 mutations. We will be using the following datasets, which we will obtain from FireBrowse:
BLCA.illuminahiseq_rnaseqv2-RSEM_genes_normalized.tar.gz: This is a zipped file containing the normalized RSEM values for bladder cancer Illumina hi-seq RNA-seq data.
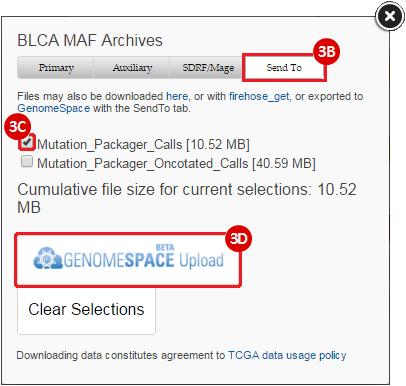
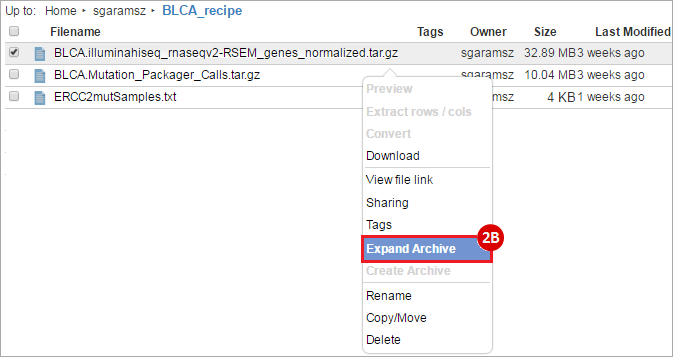
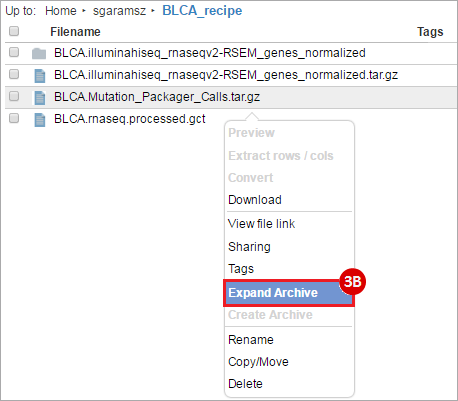
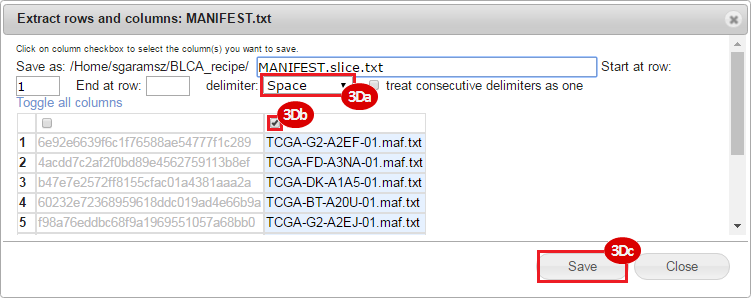
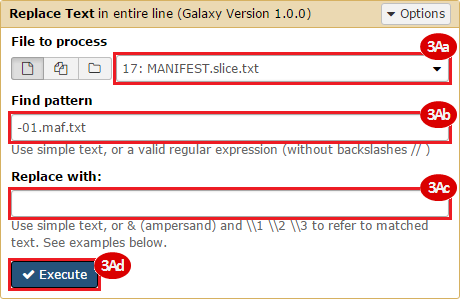
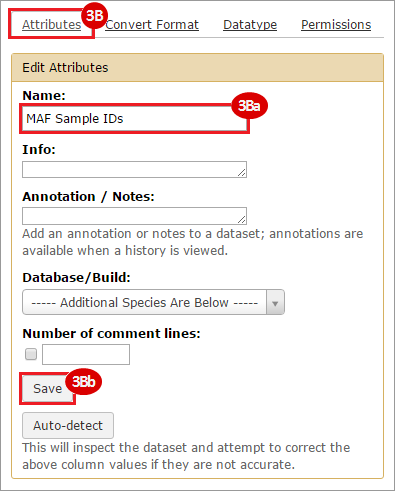
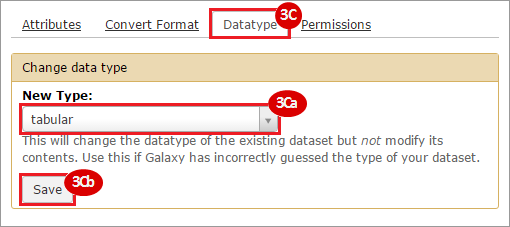
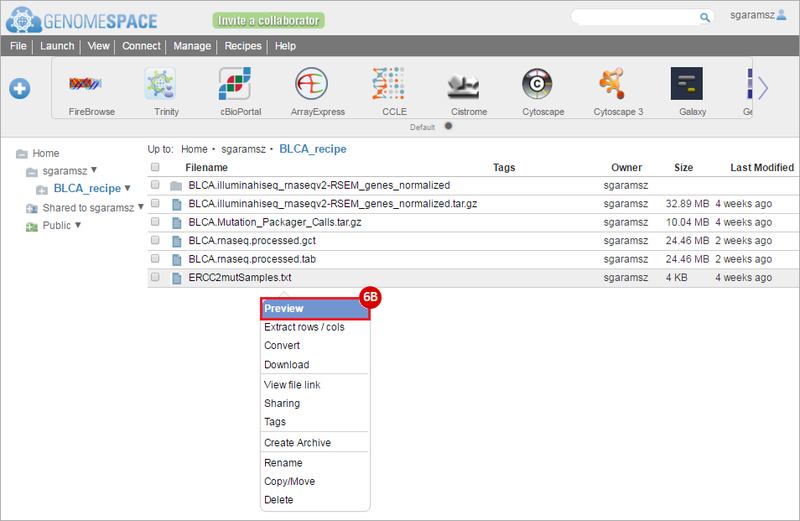
BLCA.Mutation_Packager_Calls.tar.gz: This is a zipped archive of mutation annotation files (MAFs) for all bladder cancer samples with mutations. Specifically, we will only be using the MANIFEST.txt file within this zipped archive, which just lists the samples with MAFs.
ERCC2mutSamples.txt: This text file lists the bladder cancer samples that have mutations in the ERCC2 gene. This file lists the specific type of mutation that occurs in the sample and whether it causes a missense or silent mutation of the protein product. This file also contains samples which do not have RNA-seq data, and must be filtered out.

 GenomeSpace.org
GenomeSpace.org