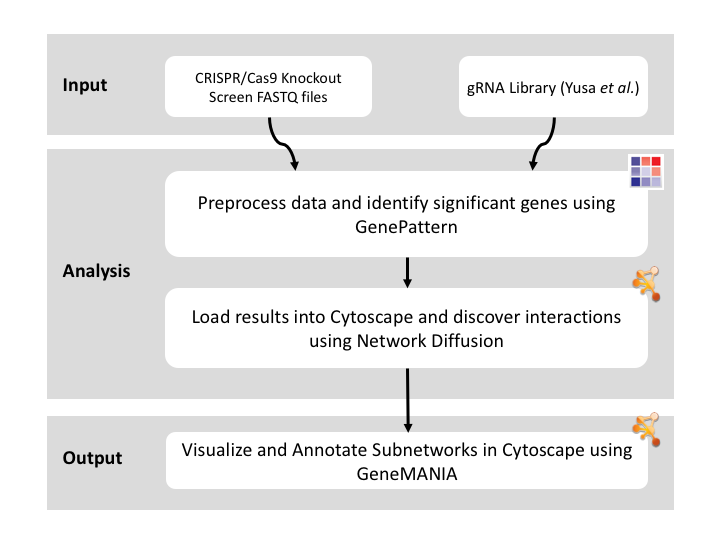
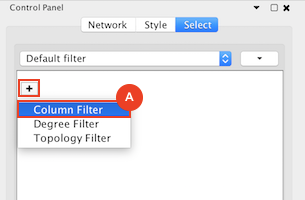
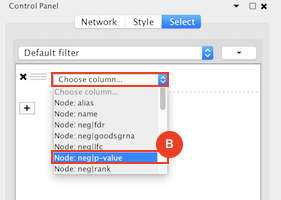
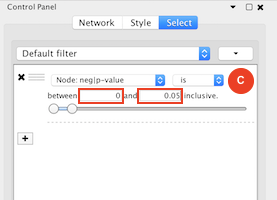
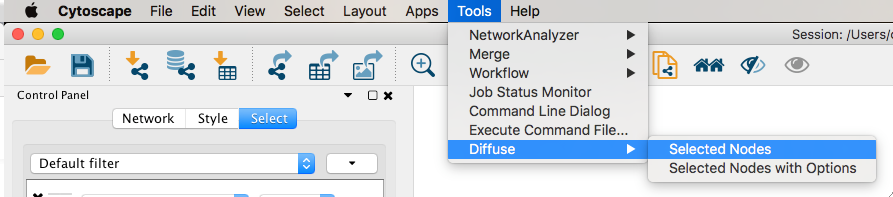
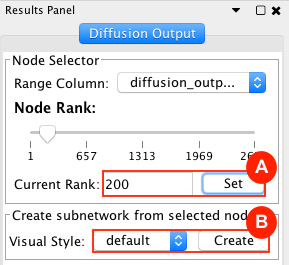
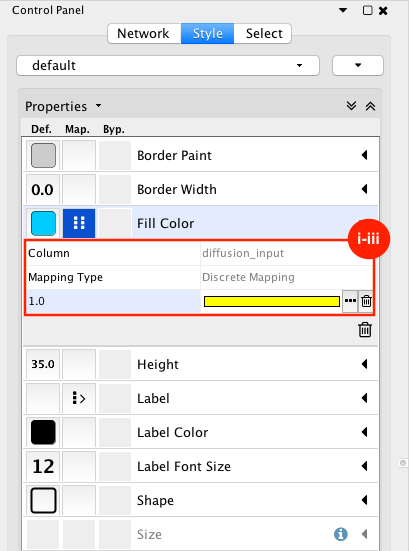
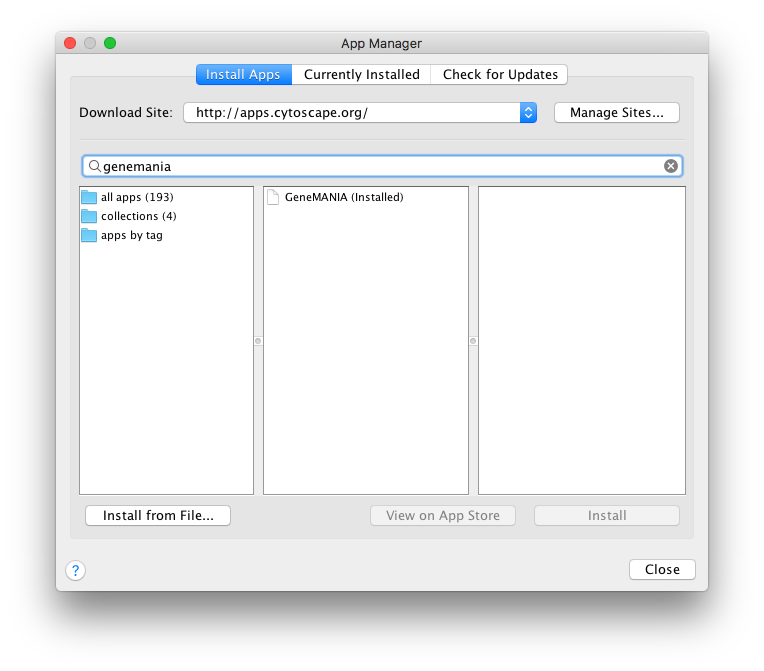
Given sgRNA read counts for our treatment and control, we use the MAGeCK algorithm on GenePattern to determine essential genes, negatively selected genes needed for ESCs proliferation in the pressence of alpha toxin. Cytoscape then allows us to understand network interactions between them. We use network diffusion to discover modules that the essential genes interact with. With the resulting subnetworks, GeneMANIA provides functional annotation by searching across a large catalogue of gene sets to understand the known processes that are enriched.
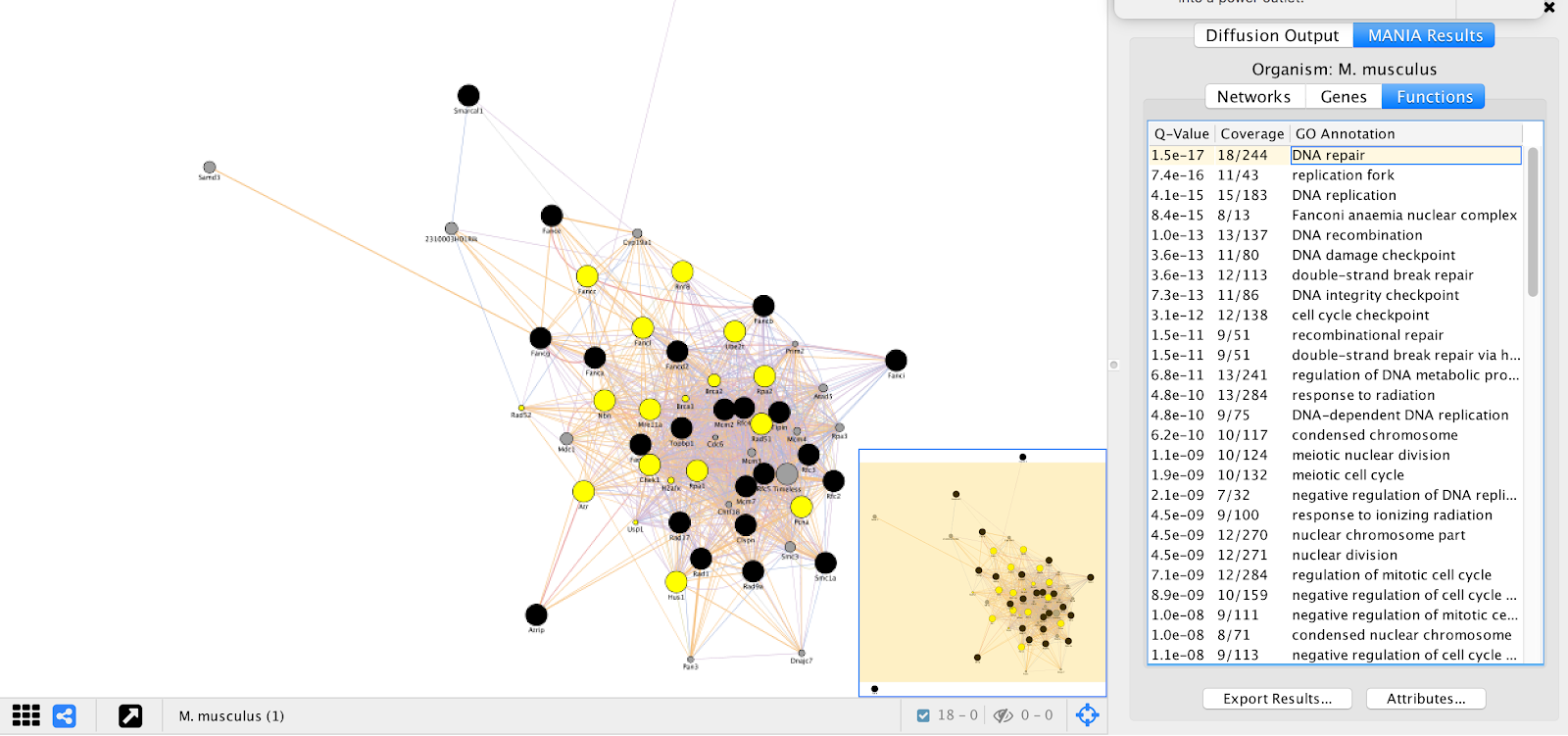
GeneMANIA provides a set of networks, genes, and functional annotations from its analysis that we can explore to understand. We can see that many of the top “Functions” listed are essential biological processes and include many DNA repair genes, which are essential for ESC proliferation in the pressence of alpha toxin. These results are consistent with results found in Koike-Yusa et. al. 2014.

 GenomeSpace.org
GenomeSpace.org