Does my RNA-Seq data contain noticeable batch effects? Can I remove any batch effects I find?
This recipe provides one method to assess RNA-seq count data for potential batch effects. Identified batch effects are removed using the ComBat algorithm (Johnson, Rabinovic, and Li). An example use case of this recipe is when an investigator wants to correct data and account for confounding variables generated by effects such as using different reagents or processing samples at different times.
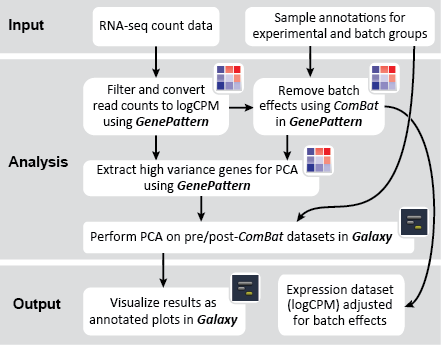
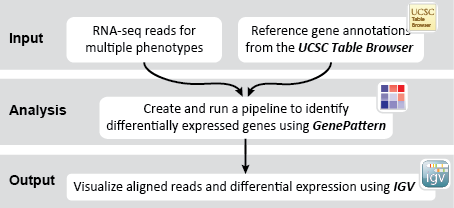
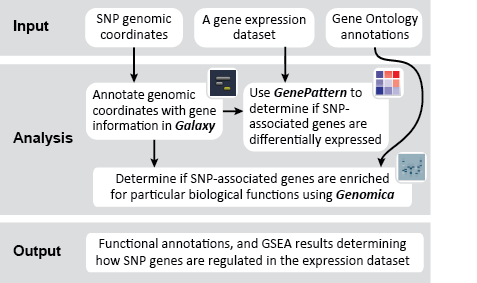
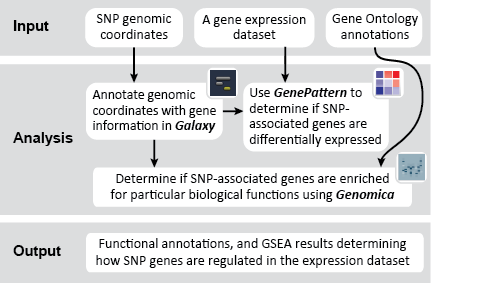
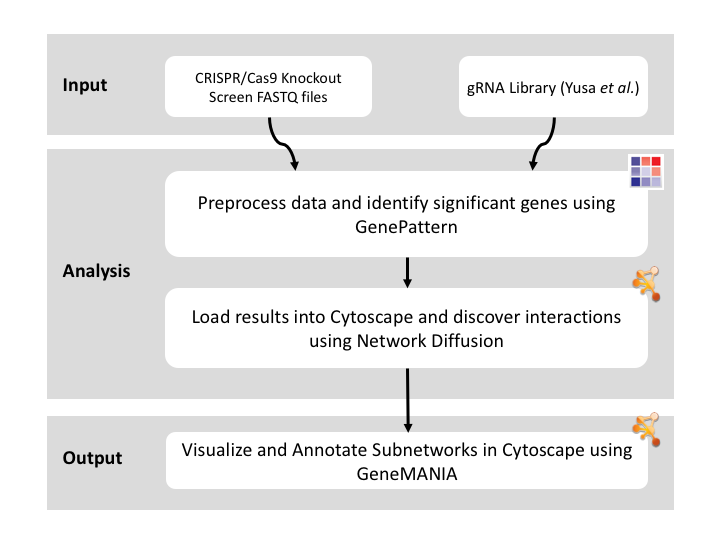
This recipe uses count data and sample annotations derived from two separate RNA-seq experiments and relies upon Principal Component Analysis (PCA) as a primary means of evaluating the dataset for batch effects. In particular, this recipe utilizes a number of GenePattern modules to filter and preprocess the RNA-seq count data and to normalize the dataset using the ComBat algorithm. We then use Galaxy to view the influence of batch effects in our pre- and post-normalization datasets.
Why consider batch effects (a.k.a. confounders)? A "batch" or "confounding" variable can be generally defined as an extraneous variable that may correlate or anti-correlate with our variables of interest. Failing to account for confounding variables or batch effects can lead us to draw spurious conclusions from our data. For example:
“Albert is comparing the hardiness of two different E. coli strains, X and Y, by measuring their production of various stress proteins under a mild bactericide. However, Albert's bactericide stock is running low and he ends up using two different stocks of the same bactericide over the course of this experiment. Here, bactericide stock is a confounding or batch variable. Perhaps one bactericide stock is more potent than the other. Albert will need to be careful in how he analyzes his data. He must somehow ensure that differences in stress protein abundances between his X and Y strains are driven by the biological differences of these two strains rather than differences in the bactericide stocks.”
Efforts can be undertaken to minimize batch effects by ensuring proper experimental design, e.g., simplifying protocols to eliminate potential confounders. If batches are unavoidable, it is also important to randomize samples across these batches such that differences between batches can be attributed to batch effects rather than biological differences. But despite these efforts, unforeseen batch effects may still be present in any experiment. Thus, we should be aware of the degree to which these unwanted factors influence our results and be prepared to use various statistical methods to remove these unwanted factors if needed.
How do we identify batch effects? There are a variety of methods for identifying batch effects. Sometimes, potential batches can be identified at the outset of a project based on its experimental design. For example, if samples must be processed in separate groups due to the limiting capacity of a sequencer, then these processing groups are likely to introduce batch effects. If batches can be pre-identified, then the scientist can include identical control samples in each batch. Any variation in measurements from these control samples can then be attributed to and used to quantify a batch effect.
Another common method is to visualize data using a PCA projection. PCA uses a linear transformation to transform a dataset into a space of linearly uncorrelated, orthogonal "principal components". More simply, PCA is a way of visualizing data such that underlying structures are revealed. We encourage users to learn more about PCA (Ma and Dai's 2011 article in Briefings in Bioinformatics is a good starting point). By re-projecting with PCA, we can identify those variables, including batch variables, that are contributing to large variances in the dataset.

 GenomeSpace.org
GenomeSpace.org